Résumés
Résumé
Une description phénoménologique précise de la différenciation de la touffe ciliaire des cellules sensorielles de l’oreille interne a été réalisée il y a environ 20 ans chez le poulet. Pourtant, les mécanismes cellulaires concourant à la formation de cette structure remarquablement organisée, qui assure la transformation d’un son ou d’une accélération en un signal électrique (transduction mécano-électrique), demeurent très mal connus. Récemment, par des approches génétiques conjointes chez l’homme et la souris, cinq des protéines directement impliquées dans le syndrome de Usher de type I, un double déficit sensoriel qui concerne à la fois l’audition et la vision, ont pu être caractérisées. Ces découvertes ont d’ores et déjà permis d’identifier les liens interstéréociliaires comme des acteurs essentiels de la différenciation des touffes ciliaires.
Summary
Defects in myosin VIIa, the PDZ-domain-containing protein harmonin, cadherin 23, protocadherin 15, and the putative scaffolding protein sans, underlie five genetic forms of Usher syndrome type I (USH1), the most frequent cause of hereditary deafness-blindness in humans. Mice mutants defective for any of these proteins have a severe hearing impairment and display similar inner ear phenotypes characterized by the abnormal spreading of the sensory cells’ stereocilia. These are highly specialized mechanoreceptive organelles derived from microvilli, that normally form a well-structured hair bundle at the apex of inner ear sensory cells. All the USH1 proteins, except sans, have been detected in the growing stereocilia. Moreover, biochemical studies have started to unravel the multiple direct molecular interactions between USH1 proteins. In particular, harmonin can bind to the other four USH1 proteins and to F-actin. Finally, cell biology studies have provided the first insights into the functions of these proteins, and revealed that cadherin 23, and probably protocadherin 15 also, are associated with transient lateral links that interconnect growing stereocilia. These connectors play a critical role in the differentiating hair bundle.
Corps de l’article
Le syndrome de Usher (USH) est la cause la plus fréquente de surdité-cécité héréditaire. Son incidence est d’environ 1/25 000. On distingue trois formes cliniques selon la sévérité de l’atteinte auditive, l’existence éventuelle d’un déficit vestibulaire associé et l’âge d’apparition de l’atteinte rétinienne (rétinopathie pigmentaire). Le syndrome de Usher de type 1 (USH1) est la forme la plus sévère. Il est caractérisé par une surdité congénitale profonde associée à une atteinte vestibulaire et par l’apparition prépubertaire de la rétinopathie pigmentaire [1, 2]. L’identification récente de plusieurs gènes responsables de USH1, ainsi que l’analyse histopathologique des différentes souris mutantes déficientes pour les protéines codées par ces gènes, ont éclairé certains des mécanismes cellulaires et moléculaires impliqués dans le développement de la touffe ciliaire des cellules sensorielles de l’oreille interne (cellules ciliées).
Les formes génétiques du syndrome USH1
Les analyses de ségrégation de marqueurs polymorphes de l’ADN effectuées dans des familles comportant plusieurs sujets atteints du syndrome USH1 ont permis d’établir l’existence d’au moins sept formes génétiques de ce syndrome. Les loci chromosomiques correspondants sont notés USH1A-G. Les gènes impliqués dans les formes USH1B, C, D, F et G ont déjà pu être identifiés par diverses stratégies de clonage positionnel[1]. Ils codent respectivement pour la myosine VIIa, une myosine non conventionnelle, l’harmonine, qui contient plusieurs domaines de type PDZ dont on sait qu’ils sont présents dans des protéines sous-membranaires organisatrices de complexes moléculaires, la cadhérine 23 et la protocadhérine 15, deux protéines transmembranaires de la famille des cadhérines, particulières par la grande taille de leur région extracellulaire et une petite protéine cytosolique, « Sans », comportant des domaines de type ankyrine (Figure 1) [1, 3]. L’analyse des transcrits issus des différents gènes USH1 dans l’oreille interne et la rétine de souris a révélé l’existence d’épissages alternatifs, suggérant la présence de plusieurs isoformes protéiques pour la plupart de ces gènes [2]. Une mutation n’affectant que certaines isoformes pourrait, dans certains cas, rendre compte du phénotype moins sévère (surdité sans rétinopathie) observé dans quelques familles [1, 2].
Figure 1
Les cinq protéines USH1 connues.
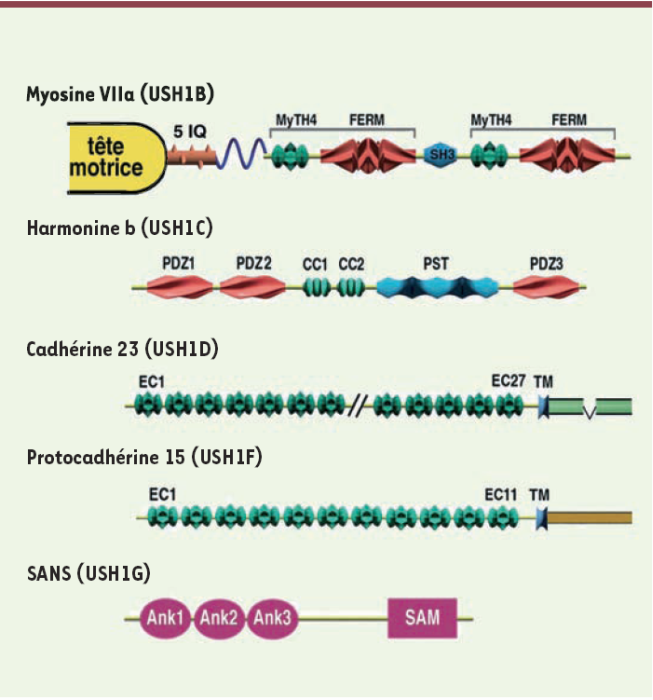
La myosine VIIa (USH1B) est une myosine non conventionnelle de 2215 acides aminés. Elle est constituée d’une tête motrice qui contient les domaines de liaison à l’ATP et à l’actine, d’une région intermédiaire contenant cinq motifs IQ (isoleucine-glutamine) qui se lient à la calmoduline, et d’une longue partie caudale (1360 acides aminés). Cette dernière contient un petit domaine d’homodimérisation (coiled coil) suivi de deux grands fragments de structure semblable, formés chacun d’un domaine MyTH4 (myosin tail homology 4) et d’un domaine FERM (4.1, ezrin, radixin, moesin) séparés par un domaine SH3 (src homology-3). Ces domaines peuvent interagir avec de nombreuses molécules [2], dont une protéine transmembranaire ubiquitaire des jonctions d’adhérence intercellulaires, la vézatine, qui participerait également à la constitution de certains liens interstéréociliaires transitoires (liens basaux) [17]. L’harmonine (USH1C, 910 acides aminés dans sa forme longue) contient trois domaines de type PDZ (initialement décrits dans les protéines PSD, Dlg-A et ZO-1, d’où leur nom), deux domaines coiled coil (CC1 et 2) et un domaine riche en proline, sérine et thréonine (PST). La cadhérine 23 (USH1D, 3354 acides aminés) comporte 27 domaines extracellulaires du type cadhérine (EC), un domaine transmembranaire (TM) et un domaine intracellulaire de 268 acides aminés sans homologie avec aucune protéine connue. La protocadhérine 15 (USH1F, 1955 acides aminés) comporte 11 domaines extracellulaires du type cadhérine (EC), un domaine transmembranaire (TM) et un domaine intracellulaire de 559 acides aminés contenant deux régions riches en proline qui constituent des domaines classiques d’interaction avec d’autres protéines. Enfin, la petite protéine (460 acides aminés), responsable de la forme USH1G, a été dénommée SANS (scaffold ankyrin and sterile alpha motif containing protein) car elle contient trois motifs de type ankyrine (Ank) ainsi qu’un motif sterile alpha (SAM), initialement décrit dans plusieurs protéines impliquées dans la différenciation sexuelle de la levure.
Des souris mutantes pour chaque gène USH1 connu
Il existe des souris porteuses de mutations dans les gènes orthologues de chacun des cinq gènes USH1 connus actuellement : souris shaker-1 (déficientes en myosine VIIa), deaf circler (harmonine), waltzer (cadhérine 23), Ames waltzer (protocadhérine 15) et jackson shaker (protéine Sans) [2]. Toutes ces souris mutantes ont un déficit auditif associé à des troubles de l’équilibre par dysfonctionnement vestibulaire. En revanche, aucune ne semble avoir d’atteinte visuelle, à la différence de ce que l’on observe chez l’homme. Seules des anomalies mineures de l’électrorétinogramme ont pu être mises en évidence chez des souris shaker-1 ou waltzer [4]. L’analyse morphologique, en microscopie électronique à balayage, de la face apicale des cellules sensorielles de l’oreille interne a révélé, chez tous les mutants murins, une désorganisation des touffes ciliaires (Figure 2). L’anomalie était présente dès le 18e jour du développement embryonnaire[2] chez les mutants shaker-1 et waltzer pour lesquels une étude chronologique a été réalisée [5].
Figure 2
Désorganisation de la touffe ciliaire des cellules sensorielles auditives chez une souris mutante shaker-1.

Figure 2 (suite)

La face apicale des cellules est vue en microscopie électronique à balayage. On note, chez une souris normale (A), l’ordonnancement remarquable « en V » des stéréocils formant la touffe ciliaire. Chez la souris shaker-1(B), déficiente en myosine VIIa, les stéréocils ont, au contraire, une disposition éparpillée (d’après [18]).
Différenciation de la touffe ciliaire des cellules sensorielles de l’oreille interne : un processus séquentiel complexe
La touffe ciliaire coiffe la surface apicale des cellules sensorielles cochléaires et vestibulaires qui sont appelées pour cette raison, cellules ciliées. Elle joue un rôle essentiel dans la transduction mécano-électrique, un processus par lequel l’énergie mécanique véhiculée par l’onde sonore ou une accélération (linéaire ou angulaire) est transformée en un signal électrique (potentiel de récepteur) dans la cellule sensorielle. Ce signal est ensuite transmis au cerveau par des neurones auditifs ou vestibulaires primaires. La touffe ciliaire est composée de 20 à 300 microvillosités rigides que l’on nomme à tort stéréocils. Comme dans toute microvillosité, les filaments d’actine des stéréocils sont uniformément polarisés (extrémité barbée située à l’extrémité apicale). Cependant, les stéréocils diffèrent des autres microvillosités par plusieurs aspects : (1) ils sont plus larges et plus longs et contiennent jusqu’à 3 000 filaments d’actine dans certaines espèces ; (2) ils se rétrécissent à leur base, juste au dessus du point d’insertion dans la membrane apicale de la cellule ; (3) ils sont disposés en trois à cinq rangées de hauteurs croissantes, la rangée la plus haute étant la plus proche de l’unique cil véritable (contenant des microtubules), appelé kinocil, qui définit l’axe de polarité planaire ; (4) ils sont reliés entre eux par différents types de liens interstéréociliaires, classés selon leur position le long des stéréocils et leurs propriétés biochimiques. Tous ces liens sont répartis plus ou moins uniformément autour de la circonférence du stéréocil, à l’exception d’un lien apical (tip link), unique, qui s’étend de l’extrémité de chaque stéréocil à la face latérale du stéréocil adjacent appartenant à la rangée de taille supérieure (Figure 3A). Les liens apicaux seraient physiquement liés aux canaux cationiques de la transduction mécano-électrique ; leur mise en tension, lors de la déflexion de la touffe ciliaire provoquée par l’onde sonore ou une accélération, entraînerait l’ouverture de ces canaux [6].
Figure 3

A. Touffe ciliaire d’une cellule sensorielle auditive immature avec ses liens interstéréociliaires.Le kinocil, structure organisée autour d’un squelette de microtubules issu d’un corps basal, est présent durant toute la période de différenciation de la touffe ciliaire. Il n’existe plus dans les cellules sensorielles de l’organe auditif mature, tandis qu’il persiste dans celles des organes vestibulaires. Trois stéréocils appartenant à trois rangées différentes sont représentés. Il s’agit en fait de microvillosités contenant de très nombreux filaments d’actine, dont quelques uns se prolongent dans le corps cellulaire pour s’ancrer dans la plaque cuticulaire, un enchevêtrement dense de filaments d’actine proche de la surface apicale de la cellule. Différents groupes de liens interstéréociliaires sont étagés depuis la base jusqu’au sommet des stéréocils. De plus, un lien apical unique (tip link) unit le sommet de chaque stéréocil à la face latérale du stéréocil adjacent de taille supérieure. Le lien apical joue un rôle essentiel dans l’ouverture du canal ionique assurant la transduction mécano-électrique. Ce canal, possiblement unique dans chaque stéréocil, serait situé au voisinage immédiat de l’une ou de l’autre extrémité du lien apical. La mise en tension du lien apical lors de la déflexion de la touffe ciliaire en direction du kinocil augmenterait la probabilité d’ouverture du canal. On note également la présence de liens unissant le kinocil aux stéréocils de grande taille voisins. Les jonctions d’adhérence entre la cellule sensorielle et les cellules de soutien adjacentes sont indiquées par des flèches. B. Immuno-détection de la cadhérine 23 dans la touffe ciliaire en microscopie électronique. Sur ces coupes longitudinale (à gauche) et transversale (à droite) de la touffe ciliaire d’une cellule sensorielle auditive de souris dans les premiers jours suivant la naissance, l’accumulation des grains d’or dans la région distale de la touffe ciliaire, indiquant la présence de la cadhérine 23, coïncide avec des liens interstéréociliaires (têtes de flèche) (barre d’échelle : 200 nm) (d’après [11]).
Les étapes du développement de la touffe ciliaire ont été décrites par Tilney et al. dans les cellules sensorielles de l’organe auditif du poulet [7]. Le premier stade est l’émergence de petites microvillosités de taille similaire, groupées autour d’un kinocil central. La migration du kinocil vers la périphérie est suivie d’une élongation séquentielle des microvillosités aboutissant à la formation de plusieurs rangées de stéréocils de taille décroissante. Tandis que les stéréocils, qui ont cessé de croître en longueur, s’élargissent, quelques uns de leurs filaments d’actine centraux s’étendent en direction du corps cellulaire et pénètrent dans un entrelacs de filaments d’actine qui formera ensuite la plaque cuticulaire, située dans un plan grossièrement parallèle à la surface apicale de la cellule (Figure 3A). Enfin, les stéréocils en excès, qui n’ont pas été incorporés dans la touffe ciliaire, régressent. Malgré certaines différences, le développement des touffes ciliaires dans l’organe auditif des mammifères s’effectue selon la même séquence. Fait notable, les stéréocils sont, dès le stade initial de leur croissance différentielle, connectés entre eux et au kinocil par de nombreux liens fibreux, qui forment un réseau plus dense que dans la touffe ciliaire mature [8].
Rôle des protéines USH1 dans le maintien de la cohésion de la touffe ciliaire en croissance
L’analyse en immunohistofluorescence, chez la souris, des différentes protéines impliquées dans le syndrome USH1 a montré, qu’à l’exception de la protéine sans, détectée dans la région apicale du corps des cellules sensorielles et le kinocil, toutes sont présentes dans les stéréocils de la touffe ciliaire au cours de sa différenciation. Alors que la myosine VIIa semble distribuée de manière assez uniforme dans la touffe ciliaire (comme d’ailleurs dans le corps cellulaire), l’harmonine, la cadhérine 23 (Figure 3B) et la protocadhérine 15 sont concentrées dans la région distale des stéréocils en croissance. La cadhérine 23, et probablement la protocadhérine 15, participent à la constitution de liens interstéréociliaires (et interkino-stéréociliaires) transitoires, différents du lien apical [9-11]. Quant à l’harmonine, dont nous avons montré qu’elle interagit directement avec l’actine filamenteuse [12], elle est vraisemblablement impliquée dans l’ancrage de ces deux molécules transmembranaires au squelette microfilamenteux des stéréocils. Par ses domaines PDZ, l’harmonine peut en effet se lier à ces protéines qui, contrairement aux cadhérines classiques, ne possèdent pas le motif consensuel de liaison à la β-caténine (une protéine cytosolique assurant l’ancrage des autres cadhérines au cytosquelette d’actine) [13]. En fait, l’harmonine est capable de se lier aux quatre autres protéines USH1 connues à ce jour [12, 14, 15], ce qui concorde avec le rôle d’organisateur de complexes moléculaires classiquement attribué aux protéines à domaines PDZ [16]. La myosine VIIa est une myosine non conventionnelle dont la longue queue modulaire (Figure 1) est capable de se lier à diverses protéines [2]. Elle pourrait jouer à la fois un rôle de transporteur moléculaire (en particulier pour l’harmonine) et localement un rôle de tenseur entre la membrane plasmique et le cytosquelette. Enfin, la protéine Sans pourrait également être impliquée dans le transport cellulaire d’autres protéines USH1 vers les stéréocils.
Conclusions
Les travaux récents concernant les protéines impliquées dans le syndrome USH1 ont mis en lumière le rôle essentiel de liens interstéréociliaires précoces et transitoires durant la croissance de la touffe ciliaire des cellules sensorielles de l’oreille interne. La participation probable de la plaque cuticulaire et des jonctions intercellulaires dans ce processus morphogénétique reste à évaluer. Certaines protéines USH1, notamment la myosine VIIa, pourraient d’ailleurs également intervenir à ce niveau.
Parties annexes
Notes
-
[1]
Voir Hereditary hearing loss homepagehttp://webhost.ua.ac.be/hhh/
-
[2]
Contrairement à l’oreille interne humaine, dont la différenciation est achevée avant la naissance, celle de la souris poursuit son développement jusqu’à la fin de la 3e semaine post-natale.
Références
- 1. Petit C. Usher syndrome : from genetics to pathogenesis. Annu Rev Genomics Hum Genet 2001 ; 2 : 271-97.
- 2. El-Amraoui A, Petit C. Usher I syndrome : unravelling the mechanisms that underlie the cohesion of the growing hair bundle in inner ear sensory cells. J Cell Sci 2005 (sous presse).
- 3. Weil D, El-Amraoui A, Masmoudi S, et al. Usher syndrome type I G (USH1G) is caused by mutations in the gene encoding SANS, a protein that associates with the USH1C protein, harmonin. Hum Mol Genet 2003 ; 12 : 463-71.
- 4. Libby RT, Steel KP. Electroretinographic anomalies in mice with mutations in Myo7a, the gene involved in human Usher syndrome type 1B. Invest Ophthalmol Vis Sci 2001 ; 42 : 770-8.
- 5. Holme RH, Steel KP. Stereocilia defects in waltzer (Cdh23), shaker1 (Myo7a) and double waltzer/shaker1 mutant mice. Hear Res 2002 ; 169 : 13-23.
- 6. Hudspeth AJ, Choe Y, Mehta AD, Martin P. Putting ion channels to work : mechanoelectrical transduction, adaptation, and amplification by hair cells. Proc Natl Acad Sci USA 2000 ; 97 : 11765-72.
- 7. Tilney LG, Tilney MS, DeRosier DJ. Actin filaments, stereocilia, and hair cells : how cells count and measure. Annu Rev Cell Biol 1992 ; 8 : 257-74.
- 8. Goodyear RJ, Marcotti W, Kros CJ, Richardson GP. Development and properties of stereociliary link types in hair cells of the mouse cochlea. J Comp Neurol 2005 ; 485 : 75-85.
- 9. Siemens J, Lillo C, Dumont RA, et al. Cadherin 23 is a component of the tip link in hair-cell stereocilia. Nature 2004 ; 428 : 950-5.
- 10. Lagziel A, Ahmed ZM, Schultz JM, et al. Spatiotemporal pattern and isoforms of cadherin 23 in wild type and waltzer mice during inner ear hair cell development. Dev Biol 2005 ; 280 : 295-306.
- 11. Michel V, Goodyear RJ, Weil D, et al. Cadherin 23 is a component of the sensory hair bundle’s transient lateral links in the developing cochlea. Dev Biol 2005 ; 280 : 281-94.
- 12. Boëda B, El-Amraoui A, Bahloul A, et al. Myosin VIIa, harmonin and cadherin 23, three Usher I gene products that cooperate to shape the sensory hair cell bundle. Embo J 2002 ; 21 : 6689-99.
- 13. Bershadsky A. Magic touch : how does cell-cell adhesion trigger actin assembly ? Trends Cell Biol 2004 ; 14 : 589-93.
- 14. Siemens J, Kazmierczak P, Reynolds A, et al. The Usher syndrome proteins cadherin 23 and harmonin form a complex by means of PDZ-domain interactions. Proc Natl Acad Sci USA 2002 ; 99 : 14946-51
- 15. Adato A, Michel V, Kikkawa Y, et al. Interactions in the network of Usher syndrome type 1 proteins. Hum Mol Genet 2005 ; 14 : 347-56.
- 16. Hung AY, Sheng M. PDZ domains : structural modules for protein complex assembly. J Biol Chem 2002 ; 277 : 5699-702.
- 17. Kussel-Andermann P, El-Amraoui A, Safieddine S, et al. Vezatin, a novel transmembrane protein, bridges myosin VIIA to the cadherin-catenins complex. EMBO J 2000 ; 19 : 6020-9.
- 18. Self T, Mahony M, Fleming J, et al. Shaker-1 mutations reveal roles for myosin VIIA in both development and function of cochlear hair cells. Development 1998 ; 125 : 557-66.
Liste des figures
Figure 1
Les cinq protéines USH1 connues.
La myosine VIIa (USH1B) est une myosine non conventionnelle de 2215 acides aminés. Elle est constituée d’une tête motrice qui contient les domaines de liaison à l’ATP et à l’actine, d’une région intermédiaire contenant cinq motifs IQ (isoleucine-glutamine) qui se lient à la calmoduline, et d’une longue partie caudale (1360 acides aminés). Cette dernière contient un petit domaine d’homodimérisation (coiled coil) suivi de deux grands fragments de structure semblable, formés chacun d’un domaine MyTH4 (myosin tail homology 4) et d’un domaine FERM (4.1, ezrin, radixin, moesin) séparés par un domaine SH3 (src homology-3). Ces domaines peuvent interagir avec de nombreuses molécules [2], dont une protéine transmembranaire ubiquitaire des jonctions d’adhérence intercellulaires, la vézatine, qui participerait également à la constitution de certains liens interstéréociliaires transitoires (liens basaux) [17]. L’harmonine (USH1C, 910 acides aminés dans sa forme longue) contient trois domaines de type PDZ (initialement décrits dans les protéines PSD, Dlg-A et ZO-1, d’où leur nom), deux domaines coiled coil (CC1 et 2) et un domaine riche en proline, sérine et thréonine (PST). La cadhérine 23 (USH1D, 3354 acides aminés) comporte 27 domaines extracellulaires du type cadhérine (EC), un domaine transmembranaire (TM) et un domaine intracellulaire de 268 acides aminés sans homologie avec aucune protéine connue. La protocadhérine 15 (USH1F, 1955 acides aminés) comporte 11 domaines extracellulaires du type cadhérine (EC), un domaine transmembranaire (TM) et un domaine intracellulaire de 559 acides aminés contenant deux régions riches en proline qui constituent des domaines classiques d’interaction avec d’autres protéines. Enfin, la petite protéine (460 acides aminés), responsable de la forme USH1G, a été dénommée SANS (scaffold ankyrin and sterile alpha motif containing protein) car elle contient trois motifs de type ankyrine (Ank) ainsi qu’un motif sterile alpha (SAM), initialement décrit dans plusieurs protéines impliquées dans la différenciation sexuelle de la levure.
Figure 2
Désorganisation de la touffe ciliaire des cellules sensorielles auditives chez une souris mutante shaker-1.
Figure 2 (suite)
La face apicale des cellules est vue en microscopie électronique à balayage. On note, chez une souris normale (A), l’ordonnancement remarquable « en V » des stéréocils formant la touffe ciliaire. Chez la souris shaker-1(B), déficiente en myosine VIIa, les stéréocils ont, au contraire, une disposition éparpillée (d’après [18]).
Figure 3
A. Touffe ciliaire d’une cellule sensorielle auditive immature avec ses liens interstéréociliaires.Le kinocil, structure organisée autour d’un squelette de microtubules issu d’un corps basal, est présent durant toute la période de différenciation de la touffe ciliaire. Il n’existe plus dans les cellules sensorielles de l’organe auditif mature, tandis qu’il persiste dans celles des organes vestibulaires. Trois stéréocils appartenant à trois rangées différentes sont représentés. Il s’agit en fait de microvillosités contenant de très nombreux filaments d’actine, dont quelques uns se prolongent dans le corps cellulaire pour s’ancrer dans la plaque cuticulaire, un enchevêtrement dense de filaments d’actine proche de la surface apicale de la cellule. Différents groupes de liens interstéréociliaires sont étagés depuis la base jusqu’au sommet des stéréocils. De plus, un lien apical unique (tip link) unit le sommet de chaque stéréocil à la face latérale du stéréocil adjacent de taille supérieure. Le lien apical joue un rôle essentiel dans l’ouverture du canal ionique assurant la transduction mécano-électrique. Ce canal, possiblement unique dans chaque stéréocil, serait situé au voisinage immédiat de l’une ou de l’autre extrémité du lien apical. La mise en tension du lien apical lors de la déflexion de la touffe ciliaire en direction du kinocil augmenterait la probabilité d’ouverture du canal. On note également la présence de liens unissant le kinocil aux stéréocils de grande taille voisins. Les jonctions d’adhérence entre la cellule sensorielle et les cellules de soutien adjacentes sont indiquées par des flèches. B. Immuno-détection de la cadhérine 23 dans la touffe ciliaire en microscopie électronique. Sur ces coupes longitudinale (à gauche) et transversale (à droite) de la touffe ciliaire d’une cellule sensorielle auditive de souris dans les premiers jours suivant la naissance, l’accumulation des grains d’or dans la région distale de la touffe ciliaire, indiquant la présence de la cadhérine 23, coïncide avec des liens interstéréociliaires (têtes de flèche) (barre d’échelle : 200 nm) (d’après [11]).