Résumés
Résumé
Pour la quasi-totalité des classes thérapeutiques antithrombotiques actuellement utilisées et à venir, le ciblage a été initialement la conséquence d’une observation et non pas le fruit d’une recherche méthodique scientifique. Une fois la preuve de l’efficacité démontrée, le but du développement pharmaceutique a été de découvrir des familles moléculaires qui améliorent l’efficacité et les relations cinétique/dynamique. Les rares cas de développement d’une stratégie contre une cible spécifique, identifiée à partir des connaissances des mécanismes fondamentaux de la thrombogenèse, n’ont pas été des succès. La nature paraît avoir déjà exploité toutes les cibles efficaces et il semble qu’il n’est possible que de la copier en essayant de l’améliorer par quelques détails pharmacocinétiques ou galéniques attractifs. Quoiqu’il en soit, le développement de nombreuses formes moléculaires dirigées contre plusieurs cibles a lieu et la mise à disposition de molécules spécifiques des différentes cibles devrait modifier notre prise en charge des états thrombotiques chez les patients : nous allons passer d’une ère où nous cherchions à utiliser au mieux les rares thérapeutiques dont nous disposions à une ère où nous aurons à déterminer, pour chaque situation thrombotique, la meilleure cible à inhiber et le meilleur degré d’inhibition à atteindre.
Summary
New antithrombotic agents are being developed not only to improve efficacy, but also to increase safety in comparison with widely used conventional agents such as the oral anticoagulants. New anticoagulant, antiplatelet, and profibrinolytic compounds are currently under study in drug development programs, and most of those in phase II or III of development are derived from the observation of natural phenomena and merely mimic processes developed by mammalians, including humans, to avoid thrombosis, or by blood-sucking insects or animals to prevent coagulation of the blood their are feeding on. By contrast, drug candidates identified by means of rigorous research and designed to target new pathways and achieve direct and specific inhibition of factors that are presumed to play an important role in thrombogenesis have generally failed to show any benefit and sometimes even induce deleterious effects. The clinical development of new drugs, even those mimicking natural phenomena, improves our knowledge of the pathogenesis of thrombosis and sheds light, retrospectively, on previous conceptual errors. The improvement in our basic knowledge and the development of new types of drugs suggest that, in contrast to the current antithrombotic compounds that are used in a broad range of clinical settings, use of new drugs should be restricted to specific situations in which their mechanisms of action are predicted to deliver the highest medical benefit. A major obstacle resides in the fact that current drug development programs are still required to comply with long obsolete guidelines based on the characteristics of first-generation antithrombotic agents, and that do not take into account the specific mechanisms of action of new drugs. This situation should change, however, and new antithrombotic drugs should soon be able to benefit from adapted development programs that will make it possible to determine their optimal risk-benefit ratio.
Corps de l’article
Les antithrombotiques influent sur les trois processus de la thrombogenèse : la coagulation plasmatique, la réactivité plaquettaire et la fibrinolyse [1].
Molécules affectant la coagulation plasmatique
La coagulation procède de l’activation de complexes qui s’assemblent sur des surfaces cellulaires. Après une phase d’initiation, consistant en l’activation du facteur VII en VIIa par le facteur tissulaire (FT) [2], survient une amplification où les premières traces de thrombine jouent un rôle de rétro-activation qui produit du facteur Xa [3] et conduit à la propagation, au cours de laquelle la thrombine, produite de manière explosive, va affecter ses cibles cellulaires et moléculaires (Figure 1) [4]. Ce résumé permet de positionner les différentes possibilités d’anticoagulation.
Figure 1
Étapes de la coagulation.

Initiation : assemblage et activation des facteurs au niveau des surfaces phospolipidiques ; amplification : rétro-activation et production explosive de thrombine (facteur IIa) libre et de thrombine liée au caillot ; propagation : action de la thrombine sur ses cibles (formation du réseau de fibrine et activation plaquettaire).
Antivitamines K ou anticoagulants oraux
C’est la classe d’anticoagulants la plus utilisée. La découverte du dicoumarol date de 1920 : c’est le constituant de la « mort aux rats ». Il a fallu attendre les années 40 pour penser qu’en l’utilisant à la dose adaptée, il pourrait avoir un effet anticoagulant thérapeutique.
Plusieurs familles d’antivitamines K (AVK), notamment les indanediones et les coumariniques, sont aujourd’hui disponibles [5]. La warfarine est le plus prescrit des AVK dans le monde. L’acénocoumarol, largement prescrit en France, a une durée de vie plus courte que la warfarine. Son utilisation dans le but d’avoir un effet « on-off » plus rapide est un manque de compréhension de la physiologie de la coagulation et du mode d’action des AVK car l’essentiel de l’effet « on-off » des AVK n’est pas dû à la pharmacocinétique du produit, mais à la durée de vie des facteurs de coagulation. Ainsi, l’utilisation d’un AVK à durée de vie courte n’améliore pas l’effet anticoagulant, mais aggrave le risque d’instabilité thérapeutique.
La classe thérapeutique devant remplacer les AVK est constituée par les inhibiteurs directs de thrombine actifs par voie orale. La première molécule, le ximélagatran, a démontré une efficacité au moins équivalente à celle des AVK et sans contrôle biologique, permettant donc un bénéfice thérapeutique net [6]. Malheureusement, ce développement a été stoppé en raison d’une augmentation retardée des concentrations d’enzymes hépatiques, survenant chez environ 7 % des patients [7].
Des concurrents ciblant la thrombine et une nouvelle classe thérapeutique constituée par les anti-Xa directs actifs s’annoncent déjà.
Héparines
L’héparine a été découverte en 1916 [8]. Les molécules d’héparine ne sont pas actives directement (Figure 2), mais potentialisent un inhibiteur physiologique, l’antithrombine (AT), un inhibiteur des facteurs Xa et IIa. Pour améliorer la pharmacocinétique de l’héparine, les molécules natives ont été clivées afin d’obtenir les différentes héparines de bas poids moléculaire (HBPM) (énoxaparine, nadroparine, daltéparine, tinzaparine, réviparine…) [9] commercialisées en 1985. Or, plus le poids moléculaire des chaînes d’héparine diminue, plus l’effet anti-Xa augmente et plus l’effet anti-IIa diminue. Ces HBPM sont antithrombotiques et comportent un risque hémorragique moindre, d’où l’hypothèse, qui a longtemps prévalu, selon laquelle l’activité anti-Xa portait l’activité antithrombotique « préventive », tandis que l’activité anti-IIa représentait l’activité antithrombotique « curative » et portait le risque hémorragique. Cette hypothèse a renforcé l’idée que la molécule idéale serait la séquence saccharidique minimale de liaison de l’héparine à l’AT, le pentasaccharide [10]. Sa forme commerciale (fondaparinux) est apparue sur le marché en 2001. Le pentasaccharide ne potentialise que l’activité anti-Xa de l’AT, son développement a démontré qu’une molécule à effet uniquement anti-Xa était effectivement antithrombotique en thrombose veineuse [11], mais aussi en thrombose artérielle [12], et a démenti qu’une molécule exclusivement anti-Xa ne faisait pas saigner.
Figure 2
Fonctions des héparines.

Les héparines sont des inhibiteurs indirects des facteurs Xa et IIa, ainsi que des facteurs VIIa, IXa, XIa et XIIa. Elles potentialisent l’effet inhibiteur physiologique de l’antithrombine (anciennement antithrombine III). Le pentasaccharide est la plus petite fraction d’héparine capable de se lier à l’antithrombine.
Ces recherches ont ouvert la voie au développement de trois classes thérapeutiques : les pentasaccharides synthétiques, les hépariniques synthétiques et les anti-Xa synthétiques directs. Concernant la première classe, le fondaparinux a été sulfaté et méthylé pour donner l’idraparinux [13]. Ces modifications ont augmenté l’affinité pour l’AT et prolongé sa durée de vie, autorisant une seule administration par semaine. Cela expose au risque de non-contrôle de l’hémorragie. Le développement de l’idraparinux a donc été arrêté pour permettre la mise au point d'une forme dont on pourrait facilement neutraliser l’activité. Quant aux hépariniques synthétiques, la connaissance des séquences saccharidiques capables de se lier à l’AT et à la thrombine a permis la synthèse de deux chaînes saccharidiques, l’une anti-Xa et l’autre anti-IIa, reliées par une séquence intercalaire sans fonction : c’est l’hexadécasaccharide. Les premiers essais montrent une grande efficacité dans les modèles de thromboses veineuse et artérielle [14]. Enfin, deux anti-Xa synthétiques directs sont actuellement en phase III de développement.
Inhibiteurs directs de thrombine
La classe des inhibiteurs de thrombine (Figure 3) provient de l’observation des sangsues qui ont la faculté de garder liquide le sang de leur victime. La molécule responsable de cette anticoagulation est l’hirudine [15]. Elle aurait pu avoir un développement exemplaire si son mode d’action et si la physiopathologie des thromboses avaient été mieux connus. En effet, les chercheurs ne se sont pas rendu compte de tout l’intérêt de cette molécule qui, du fait de sa très forte affinité pour la thrombine et surtout pour la thrombine du caillot [16], aurait dû faire penser qu’elle pouvait être efficace avec des doses capables d’inhiber localement la thrombine du caillot sans être profondément anticoagulante au niveau systémique. Cette mauvaise compréhension des mécanismes a fait utiliser la molécule à des doses trop anticoagulantes : les effets de ce surdosage inutile ont été tels que la molécule a quasiment cessé d’être prescrite.
Figure 3
Inhibiteurs directs de thrombine (facteur IIa).
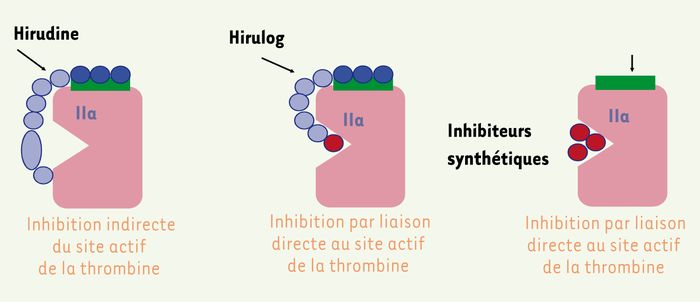
L’erreur a été alors de synthétiser des dérivés de faible affinité pour faire, pensait-on, « moins saigner », sans comprendre que, si l’affinité est diminuée, les concentrations vont devoir être augmentées. L’objectif particulièrement attrayant d’inhiber seulement la thrombine du caillot tout en préservant une fonction hémostatique systémique allait être perdu. C’est ainsi que la bivalirudine va apparaître moins affine pour la thrombine. Son développement a été lent et uniquement centré sur la thrombose coronaire, peut-être parce que le risque hémorragique est plus « acceptable » en cardiologie d’urgence [17].
Plus récemment, les inhibiteurs directs de la thrombine, actifs par voie orale, ont bénéficié d’un développement classique avec cependant, pour le ximélagatran, un soutien promotionnel exceptionnellement fort qui a eu pour effet d’attirer l’attention sur tous ses effets, y compris ses effets secondaires [18, 19] ; son hépatotoxicité a signé la mort de la première molécule de sa classe, et fait observer à la loupe les effets de son premier challenger, le dabigatran [20]. Alors que tout l’intérêt de cette classe tient dans l’affinité des molécules pour la thrombine, c’est sur leur pharmacocinétique et leur hépatotoxicité que les nouvelles molécules semblent être choisies.
Inhibiteurs directs du facteur Xa actifs par voie orale
Il existe une logique au développement des anti-Xa directs (Figure 4), en effet la formation du facteur Xa est une étape clé de la coagulation. En théorie, une régulation du facteur Xa a plus de chance d’être calibrée qu’une intervention à une étape initiale ou terminale. Cela est conforté par les résultats obtenus avec l’inhibition indirecte du facteur Xa par le fondaparinux.
C’est tout l’intérêt porté au développement actuel du BAY 59-7939 et du LY 517717 [21]. Le développement de ces molécules est « classique », c’est-à-dire une « preuve de concept » dans la condition clinique « modèle » que constitue la prévention des événements thrombo-emboliques veineux en chirurgie orthopédique [22, 23]. Les premiers résultats confortent ce qui était attendu : ces molécules ont une bonne efficacité antithrombotique, avec une absence de rapport dose/efficacité pour un large éventail de doses, une augmentation de l’incidence des hémorragies pour les plus fortes doses laissant augurer d’une large gamme de doses efficaces et sûres.
Figure 4
Inhibition directe du facteur Xa et inhibition indirecte du facteur Xa par liaison du pentasaccharide à l’antithrombine (AT).

Inhibiteurs de la phase initiale de la coagulation
Des molécules ont été développées pour s’opposer au facteur tissulaire (FT) [24]. L’inhibiteur de la voie du facteur tissulaire (TFPI), développé dans la coagulation intravasculaire disséminée (CIVD), manque d’efficacité. Quant au facteur VII préactivé puis irréversiblement inhibé, il est peu efficace ; cet échec était relativement prévisible, puisqu’en pathologie humaine, les déficits sévères en facteur VII ne sont pas totalement protégés des accidents thrombotiques et peuvent provoquer des accidents hémorragiques graves. On ne peut donc pas espérer une efficacité antithrombotique, même par inhibition quasi complète, sans risque hémorragique significatif. Enfin, le NAPc-2, un polypeptide extrait de Ancylostoma caninum qui inhibe le complexe FVIIa/FT après s’être lié au facteur X, a été évalué en phase II en chirurgie orthopédique et dans le cadre de l’angioplastie coronaire.
Molécules affectant la réactivité plaquettaire
Les membranes cellulaires des plaquettes activées forment une surface d’assemblage pour les facteurs de coagulation. Elles fixent les microparticules circulantes portant le FT qui permet de débuter la coagulation. En retour, les plaquettes sont la première cible cellulaire sanguine de la thrombine (Figure 5).
Figure 5
Inhibiteurs de l’agrégation plaquettaire.

Le thromboxane A2 et l’ADP, activateurs plaquettaires, sont les cibles respectives de l’aspirine et des thiénopyridines. De leur côté, les anti-GPIIb/IIIa empêchent l’agrégation plaquettaire en inhibant la fixation interplaquettaire du fibrinogène (Fbg).
Aspirine
Après son effet anti-inflammatoire, l’aspirine a également montré un effet anti-agrégant plaquettaire [25]. Là encore, les découvertes ont été réalisées un peu à l’envers de la rigueur cartésienne : c’est grâce à l’aspirine que le rôle du thromboxane plaquettaire (pro-agrégant) et de la prostacycline vasculaire (anti-agrégante) dans l’athérothrombose a été évalué. Progressivement, les doses d’aspirine ont été réduites pour ne pas déséquilibrer la balance thromboxane/prostacycline [26], devenant « homéopathiques » et faisant apparaître le phénomène de la résistance à l’aspirine, qui se résume en l’évidence qu’un sous-dosage entraîne une perte d’efficacité [27]. Avec la prostacycline, anti-agrégante et vasodilatatrice, on a imaginé détenir le médicament cardiovasculaire de l’avenir. Trente ans après, qu’en reste-t-il ? Des indications limitées dans l’ischémie critique [28]. C’est pour limiter les effets secondaires de l’aspirine que l’on a essayé d’inhiber d’autres cibles de la voie des thromboxanes par des inhibiteurs de la thromboxane synthase ou des antagonistes des récepteurs TP. Leur développement est difficile, car il nécessite des essais sur des dizaines de milliers de patients pour montrer des résultats supérieurs à l’aspirine, dont le très faible prix constitue également un obstacle économique important pour le développement d’autres produits.
Thiénopyridines
Initialement anti-inflammatoire, la ticlopidine a été développée comme anti-agrégant plaquettaire. Après avoir tenté d’expliquer son efficacité par un effet sur les prostaglandines, on a trouvé qu’elle inhibait la voie de l’ADP et, plus récemment, du récepteur P2Y12. Ainsi sont nés la ticlopidine, puis le clopidogrel [29] et bientôt le prasugrel [30]. Il existe une grande variabilité dans l’utilisation de la voie de l’ADP par les plaquettes. Quel degré d’inhibition faut-il alors ? Cette incertitude, associée à la variabilité du métabolisme des thiénopyridines, explique les discussions actuelles sur l’adaptation des doses et sur l’intérêt de leur surveillance biologique, et complique d’emblée le développement de nouvelles thiénopyridines inhibitrices du P2Y12 ou du P2Y1.
Ces différents inhibiteurs, appelés anti-agrégants plaquettaires, ne sont que des anti-activants plaquettaires. Les vrais anti-agrégants plaquettaires sont les anti-GP IIb/IIIa.
Anti-GP IIb/IIIa
Après la découverte des glycoprotéines des membranes plaquettaires et l’étude de la fonction de la GP IIb/IIIa grâce à un anticorps, l’idée originale a été de prouver l’efficacité de ce réactif de laboratoire dans des modèles de thrombose, puis de l’utiliser chez l’homme sous la forme de l’abciximab [31]. Les industriels ont alors reproduit l’activité de cet anticorps par d’autres molécules, comme l’eptifibatide et le tirofiban [32]. Cependant, pour que l’activité anti-agrégante plaquettaire soit efficace, l’inhibition des récepteurs GP IIb/IIIa doit être complète. Cette nécessité est déduite de l’observation de patients qui, n’ayant que 50 % des GP IIb/IIIa et ayant une hémostase normale, ne sont pas protégés contre les accidents ischémiques artériels. Bien plus, quand les récepteurs GP IIb/IIIa sont inhibés, ils transmettent un message d’activation et tous les récepteurs GP IIb/IIIa non inhibés restent fonctionnels et responsables d’un état prothrombogène. Contre toutes ces évidences, beaucoup d’industriels se sont lancés à grands frais dans le développement de molécules anti-GP IIb/IIIa actives par voie orale. Or, pour être antithrombotique, il faut inhiber la totalité des récepteurs GP IIb/IIIa et reproduire alors, volontairement, un état Glanzmann-like dont le risque et la gravité des accidents hémorragiques associés sont connus. Ce risque, acceptable à la phase initiale d’un syndrome coronaire aigu, est inacceptable pour un traitement chronique. Ces développements ont utilisé une dose réduite, croyant limiter le risque hémorragique. Le résultat a été ce que certains experts attendaient : une absence d’efficacité antithrombotique et même un risque thrombotique augmenté [33]. Une erreur conceptuelle a donc conduit à un considérable échec de développement.
Molécules agissant sur le système fibrinolytique
La fibrinolyse physiologique [34] résulte de l’activation du plasminogène en plasmine par un activateur d’origine endothéliale, le t-PA (tissue plasminogen activator), contre lequel il existe un inhibiteur physiologique, le PAI-1 (plasminogen activator inhibitor-1). Le premier thrombolytique à avoir été commercialisé est la streptokinase (SK), produite par un streptocoque hémolytique. La SK est responsable de la production de plasmine circulante, qui s’attaque non seulement aux réseaux de fibrine des thrombus, mais aussi au fibrinogène. Cette fibrinogénolyse nécessite d’être monitorée lors des traitements par la SK. Est apparue ensuite l’urokinase (UK), extraite des urines ou des cultures de cellules rénales. Comme la SK, c’est une molécule fibrinogénolytique.
La première des molécules spécifiques de la fibrine, donc moins fibrinogénolytique, a été le t-PA (altéplase), dont la très brève durée de vie implique une utilisation en perfusion [35]. Pour obtenir des thrombolytiques spécifiques de la fibrine et de durée de vie prolongée, la séquence naturelle du t-PA a été modifiée de manière à ralentir son élimination (rétéplase, ténectéplase).
Plusieurs thrombolytiques sont actuellement en développement, qui améliorent les caractéristiques de la molécule leader dans sa classe, le tPA, ou copient des activateurs naturels. Le plus étudié actuellement est la staphylokinase (SAK), sécrétée par certaines souches de staphylocoques [36]. La SAK se fixe au plasminogène d’autant plus facilement qu’il est lié à un réseau de fibrine. Les premiers résultats prouvent l’efficacité de ce mode d’action. La dose optimale doit encore être établie, mais l’origine microbienne de la SAK provoque l’apparition d’anticorps potentiellement neutralisants chez l’homme.
L’inhibition thérapeutique du PAI-1 dans le but d’exacerber l’activité fibrinolytique naturelle est une autre voie de recherche [37].
Existe-t-il des cibles non explorées ?
La thrombine est non seulement l'enzyme finale de la coagulation mais c'est aussi un activateur puissant des plaquettes par l'intermédiaire du récepteur spécifique PAR-1 présent à leur surface. Quelle sera l'efficacité antithrombotique du seul blocage des récepteurs PAR-1 ? Le premier inhibiteur spécifique des récepteurs PAR-1 est en cours de développement dans les syndromes coronaires aigus.
Les recherches s’intéressent également aux liaisons des plaquettes avec les autres cellules sanguines et vasculaires, et particulièrement à la voie de la P-sélectine et de son ligand [38]. Ce type de liaison membranaire est utilisé aussi par les plaquettes activées pour fixer les microparticules exprimant le FT. L’inhibition de cette voie est potentiellement antithrombotique.
Inhiber l’annexine V pour inhiber l’activité procoagulante des plaquettes a été une piste, mais il y en a beaucoup d’autres [39].
Conclusions
Avec ces nouvelles thérapeutiques dirigées contre des cibles spécifiques, nous allons passer d’une ère qui cherchait à utiliser au mieux les rares thérapeutiques disponibles à une ère où il faudra choisir, pour chaque situation thrombotique, la meilleure cible à inhiber et le degré d’inhibition à atteindre. Dans cette évolution fondamentale, l’industrie pharmaceutique n’a pas encore franchi le pas d’orienter le développement d’une nouvelle molécule vers le type de thrombose où elle devrait être le plus efficace. En effet, le développement de toutes les nouvelles molécules suit le même schéma d’évaluation antithrombotique en prévention de la maladie thrombo-embolique veineuse rencontrée en chirurgie orthopédique. Puis il passe au traitement curatif des événements thrombo-emboliques veineux, à la prévention des événements thrombo-emboliques de la fibrillation auriculaire et au traitement de la phase aiguë des syndromes coronariens. Ce n’est peut-être pas l’industrie pharmaceutique qui est responsable de ce schéma stéréotypé, mais les tout-puissants systèmes de régulation et d’autorisation de mise sur le marché : ils ont établi des recommandations dans un environnement thérapeutique restreint à une seule classe d’antithrombotiques à action immédiate, les hépariniques, et à une seule classe d’antithrombotiques chroniques, les AVK. L’apparition de nouvelles molécules devrait faire réagir et changer le système. Cependant, la lourdeur de ces processus administratifs régulateurs suggère que ces changements pourraient être longs à survenir.
Parties annexes
Références
- 1. Kung C, Hayes E, Mann KG. A membrane-mediated catalytic event in prothrombin activation. J Biol Chem 1994 ; 269 : 25838-48.
- 2. Butenas S, Mann KG. Kinetics of human factor VII activation. Biochemistry 1996 ; 35 : 1904-10.
- 3. Roberts HR, Hoffman M, Monroe DM. A cell-based model of thrombin generation. Semin Thromb Haemost 2006 ; 32 (suppl 1) : 32-8.
- 4. Davie EW, Kulman JD. An overview of the structure and function of thrombin. Semin Thromb Haemost 2006 ; 32 (suppl 1) : 3-15.
- 5. Ansell J, Hirsh J, Poller L, et al. The pharmacology and management of the vitamine K antagonists. The seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest 2004 ; 126 : S204-33.
- 6. Executive steering committee on behalf of the SPORTIF investigators. Stroke prevention with oral direct thrombin inhibitor ximelagatran compared with warfarin in patients with non-valvular atrial fibrillation (SPORTIF III) : randomised controlled trial. Lancet 2003 ; 362 : 1691-8.
- 7. Clement B, Lopian K. Characterization of in vitro biotransformation of new, orally active, direct thrombin inhibitor ximelagatran, an amidoxine and ester progrug. Drug Metab Disp 2003 ; 31 : 645-51.
- 8. McLean J. The thromboplastic action of cephalin. Am J Physiol 1916 ; 41 : 250-7.
- 9. Hirsh J, Raschke R. Heparin and low molecular weight heparin. The seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest 2004 ; 126 : S188-203.
- 10. Choay J, Lormeau JC, Petitou M, et al. Structural studies on a biologically active hexasaccharide obtained from heparin. Ann NY Acad Sci 1981 ; 370 : 644-9.
- 11. Blann AD, Lip GY. Venous thromboembolism. Br Med J 2006 ; 332 : 215-9.
- 12. Yusuf S, Mehta SR, Chrolavicius S, et al. Effects of fondaparinux on mortality and reinfarction in patients with ST-segment elevation myocardial infarction : the OASIS-6 randomized trial. JAMA 2006 ; 295 : 1519-30.
- 13. Koopman MM, Büller HR. Short- and long-acting synthetic pentasaccharides. J Intern Med 2003 ; 254 : 335-42.
- 14. Bal dit Sollier C, Kang C, Berge N, et al. Activity of a a synthetic hexadecasaccharide (SanOrg123781A) in a pig model of arterial thrombosis. J Thromb Haemost 2004 ; 2 : 925-30.
- 15. Markwardt F. Studies on hirudin. Arch Exp Pathol Pharmakol 1956 ; 228 : 220-1.
- 16. Weitz JI, Hudoba M, Massel D, et al. Clot-bound thrombin is protected from inhibition by heparin-antithrompbin III but is susceptible to inactivation by antithrombin III-independent inhibitors. J Clin Invest 1990 ; 86 : 385-91.
- 17. Lincoff AM, Kleiman NS, Kereiakes, et al. Long-term efficacy of bivalirudin and provisional glycoprotein IIb/IIIa blockade versus heparin and planned glycoprotein IIb/IIIa blockade during percutaneous coronary revascularieation. REPLACE-2 randomized trial. JAMA 2004 ; 292 : 696-703.
- 18. Eriksson BI, Agnelli G, Cohen AT, et al. Direct thrombin inhibitor melagatran followed by oral ximelagatran in comparison with enoxaparin for the prevention of venous thromboembolism after total hip or knee replacement. The METHRO III study. Thromb Haemost 2003 ; 89 : 288-96.
- 19. Fiessinger JN, Huisman MV, Davidson BL, et al. Ximelagatran versus low molecular weight heparin and warfarin for the treatment of deep vein thrombosis. JAMA 2005 ; 293 : 681-9.
- 20. Eriksson BI, Dahl OE, Büller HR, et al. A new oral thrombin inhibitor, dabigatran etexilate, compared with enoxaparin for the prevention of thromboembolic events following total hip and knee replacement : the BISTRO II randomized trial. J Thromb Haemost 2005 ; 3 : 103-11.
- 21. Hampton T. New oral anticoagulants show promise. JAMA 2006 ; 295 : 743-4.
- 22. Turpie AG, Fisher WD, Bauer KA, et al. BAY 59-7939: an oral, direct factor Xa inhibitor for the prevention of venous thromboembolism in patients after total knee replacement. A phase II dose-ranging study. J Thromb Haemost 2005 ; 3 : 2479-86.
- 23. Eriksson BI, Borris L, Dahl OE, et al. Oral, direct factor Xa inhibition with BAY 59-7939 for the prevention of venous thromboembolism after total hip replacement. J Thromb Haemost 2006 ; 4 : 121-8.
- 24. Houston DS. Tissue factor : a therapeutic target for thrombotic disorders. Expert Opin Ther Targets 2002 ; 6 : 159-74.
- 25. Roth GJ, Stanford N, Majerus PW. Acetylation of prostaglandin synthase by aspirin. Proc Natl Acad Sci USA 1975 ; 72 : 3073-7.
- 26. Antithrombotic Trialists Collaboration. Collaborative metaanalysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high-risk patients. Br Med J 2002 ; 324 : 71-86.
- 27. Patrono C. Aspirin resistance : definition, mechanisms and clinical read-outs. J Thromb Haemost 2003 ; 1 : 1710-3.
- 28. Fiessinger JN, Schafer M. Trial of iloprost versus aspirin treatment for critical limb ischaemia of thromboangiitis obliterans. The TAO study. Lancet 1990 ; 335 : 555-7.
- 29. Quinn MJ, Fitzgerald DJ. Ticlopidine and clopidogrel. Circulation 1999 ; 100 : 1667-72.
- 30. Wiviott SD, Antman EM, Winters KJ, et al. Randomized comparison of prasugrel (CS-747, LY640315) a novel thienopyridine P2Y12 antagonist, with clopidogrel in percutaneous coronary intervention : results of the Joint utilization of medications to block platelets optimally (JUMBO)-TIMI 26 trial. Circulation 2005 ; 111 : 3366-73.
- 31. Coller BS. Platelet GPIIb/IIIa antagonists : the first antiintegrin receptor therapeutics. J Clin Invest 1997 ; 99 : 1467-71.
- 32. Batchelor WB, Tolleson TR, Huang Y, et al. Randomized comparison of platelet inhibition with abciximab, tirofiban and eptifibatide during percutaneous coronary intervention in acute coronary syndromes : the COMPARE trial (Comparison of measurements of platelet aggregation with aggrastat, reopro, and eptifibatide). Circulation 2002 ; 106 : 1470-6.
- 33. Holmes MB, Sobel BE, Cannon CP, et al. Increased platelet reactivity in patients given orbofiban after an acute coronary syndrome : an OPUS-TIMI 16 substudy. Am J Cardiol 2000 ; 85 : 491-3.
- 34. Collen D. The plasminogen (fibrinolytic) system. Thromb Haemost 1999 ; 82 : 259-70.
- 35. Linjen HR, Collen D. Fibrinolytic agents : mechanisms of activity and pharmacology. Thromb Haemost 1995 ; 74 : 387-90.
- 36. Damaschun G, Damacshun H, Gast K, et al. Physical and conformational properties of staphylokinase in solution. Biochim Biophys Acta 1993 ; 1161 : 244-8.
- 37. Friederich P, Levi M, Biemond B, et al. Low-molecular weight inhibitor of PAI-1 (XR5118) promotes endogenous fibrinolysis and reduces postthrombolysis thrombus growth in rabbits. Circulation 1997 ; 96 : 916-21.
- 38. Romo GM, Dong JF, Schade AJ, et al. The glycoprotein Ib-IX-V complex is a platelet counter-receptor for P-selectin. J Exp Med 1999 ; 190 : 803-13.
- 39. Joop K, Berkmans RJ, Niewland R, et al. Microparticles from patients with multiple organ dysfunction syndrome and sepsis support coagulation through multiple mechanisms. Thromb Haemost 2001 ; 85 : 810-20.
Liste des figures
Figure 1
Étapes de la coagulation.
Initiation : assemblage et activation des facteurs au niveau des surfaces phospolipidiques ; amplification : rétro-activation et production explosive de thrombine (facteur IIa) libre et de thrombine liée au caillot ; propagation : action de la thrombine sur ses cibles (formation du réseau de fibrine et activation plaquettaire).
Figure 2
Fonctions des héparines.
Les héparines sont des inhibiteurs indirects des facteurs Xa et IIa, ainsi que des facteurs VIIa, IXa, XIa et XIIa. Elles potentialisent l’effet inhibiteur physiologique de l’antithrombine (anciennement antithrombine III). Le pentasaccharide est la plus petite fraction d’héparine capable de se lier à l’antithrombine.
Figure 3
Inhibiteurs directs de thrombine (facteur IIa).
Figure 4
Inhibition directe du facteur Xa et inhibition indirecte du facteur Xa par liaison du pentasaccharide à l’antithrombine (AT).
Figure 5
Inhibiteurs de l’agrégation plaquettaire.
Le thromboxane A2 et l’ADP, activateurs plaquettaires, sont les cibles respectives de l’aspirine et des thiénopyridines. De leur côté, les anti-GPIIb/IIIa empêchent l’agrégation plaquettaire en inhibant la fixation interplaquettaire du fibrinogène (Fbg).