Résumés
Résumé
Le syndrome du X fragile, première cause de retard mental héréditaire, est une maladie monogénique liée au chromosome X. Le syndrome est causé par l’inactivation du gène Fragile Mental Retardation 1(FMR1) entraînant l’absence de la protéine FMRP dont le rôle présumé est de coordonner le devenir et la traduction d’un grand nombre d’ARNm. Toutefois, s’il est actuellement admis que FMRP se comporte comme un répresseur de la traduction dans certaines conditions expérimentales, et malgré les nombreuses publications sur le sujet, nous devons nous rendre à l’évidence que les fonctions réelles de FMRP sont encore mal connues. De plus, l’existence de deux protéines FXR1P et FXR2P, homologues à FMRP, suggère que la fonction de FMRP est bien plus complexe que celle imaginée à l’origine. Nous limitons les propos de cet article à l’état actuel des connaissances concernant le rôle de FMRP dans l’adressage des ARNm, ainsi qu’aux conséquences possibles de l’absence de FMRP sur le transport et la traduction des ARNm dans les cellules pourvues d’arborescences et de prolongements que sont les neurones.
Summary
Fragile X syndrome is the most common form of inherited mental retardation. This X-linked disease is due to transcriptional silencing of the Fragile Mental Retardation 1 (FMR1) gene and the absence of its gene product, FMRP. This protein is an RNA-binding protein present in mRNP complexes associated with the translation machinery and is thought to be a key player in the control of mRNA transport in neurons. However, the exact role of FMRP in translation remains unclear. Two homologous proteins, FXR1P and FXR2P, are also found in RNP complexes containing FMRP, suggesting that FMRP’s functions are much more complex than first thought. The molecular mechanisms altered in cells lacking FMRP still remain to be elucidated, as well as the putative roles of FXR1P and FXR2P as compensatory molecules. Here, we review the various possible functions of FMRP in RNA localization and transport in highly differentiated cells containing dendritic extensions such as neurons.
Corps de l’article
« With Fragile X, we’ve got just one protein missing, so it’s a simple problem »
Sir James Watson
Fragile comme un X
Décrit par Lubs en 1969, le syndrome X fragile se manifeste par une altération des fonctions cognitives supérieures, allant de simples difficultés d’apprentissage à un retard mental sévère. Le syndrome est souvent associé à une dysmorphie faciale et/ou ligamentaire et à une macroorchidie chez les individus de sexe masculin. La maladie touche en moyenne un homme sur 4000 et une femme sur 7000 [1, 2]. Cependant, cette incidence est peut-être sous-estimée : en effet, un retard mental léger ou l’absence de manifestations physiques évidentes peuvent échapper au diagnostic clinique. Par ailleurs, celui-ci est difficile à effectuer du fait de l’hétérogénéité des troubles comportementaux (déficits d’attention, hyperactivité, autisme) qui sont des signes également associés à d’autres maladies [3].
Le gène FMR1 en cause dans la maladie a été localisé en 1991 à l’extrémité télomérique du bras long du chromosome X dans un site fragile, d’où son appellation. Ce site, présent sous la forme d’une décondensation locale du chromosome visible au microscope, est un des éléments clés du diagnostic génétique. Il s’agit d’une anomalie due à une augmentation du nombre de répétitions du triplet CGG situé dans la région 5’ non codante de FMR1. En tant que mutation, elle peut varier en longueur d’une génération à l’autre, d’une personne à une autre, et au sein d’un même individu atteint. Une personne normale présente moins de 60 répétitions ; un individu en possédant de 60 à 200 est considéré comme « prémuté ou porteur ». Le syndrome se développe lorsque le nombre de répétitions dépasse 200, entraînant, dans la majorité des cas, une hyperméthylation des îlots CpG situés en amont du gène FMR1, causant son inactivation. La protéine correspondante, FMRP, est donc absente chez ces patients [1, 2].
Un portrait de famille
FMRP fait partie d’une famille de protéines liant l’ARN appelée Fragile X Related (FXR), comprenant les paralogues Fragile X Related 1 (FXR1P) et Fragile X Related 2 (FXR2P) [4]. Les gènes FXR1 et FXR2 sont autosomiques : aucune maladie n’est associée à ces locus. Comme FMRP est en cause dans le syndrome du X fragile, la majorité des études est consacrée à cette protéine ; cependant, les trois protéines se ressemblent structurellement (Figure 1) et possèderaient des fonctions analogues susceptibles de se substituer l’une à l’autre et ainsi de compenser éventuellement un déficit fonctionnel. Les FXRP peuvent former des homo- et des hétérodimères via leur domaine d’interaction protéine-protéine (PPID) [5]. Par ailleurs, elles contiennent deux motifs KH et un domaine riche en arginine et en glycine appelé boîte RGG, caractéristiques des protéines liant l’ARN. De plus, les FXRP transitent entre le noyau et le cytoplasme grâce à leurs signaux de localisation nucléaire (NLS) et d’export nucléaire (NES) [4]. Enfin, un domaine de phosphorylation en carboxyterminal régulerait l’association de FMRP avec l’appareil de traduction [5].
Figure 1
Structure des membres de la famille FXRP et distribution de ces protéines dans divers tissus et organes de la souris adulte.
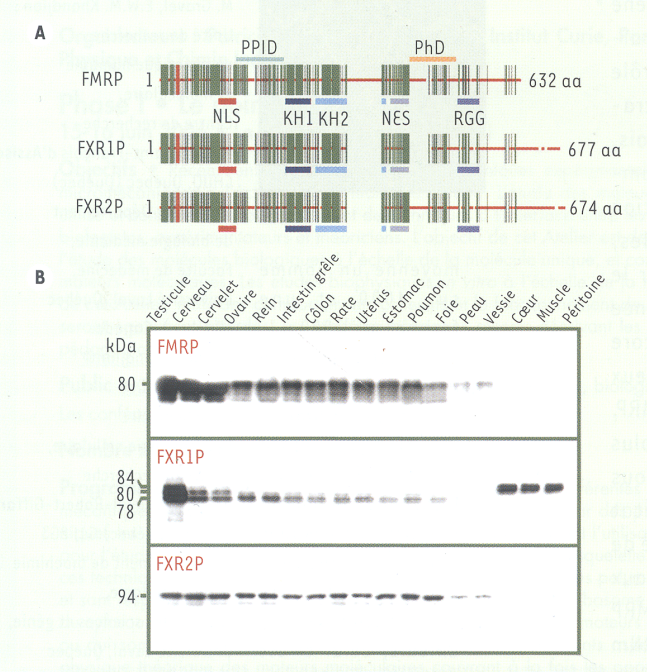
A. Structure des protéines FXR : les barres verticales (en vert foncé) correspondent aux acides aminés (aa) identiques, tandis que les barres horizontales (en rouge) représentent les aa divergents. Les domaines fonctionnels correspondant aux signaux de localisation nucléaire (NLS) et d’export nucléaire (NES), aux motifs de reconnaissance de l’ARN (KH1, KH2 et RGG), au domaine d’interaction protéine-protéine (PPID) et enfin au domaine phosphorylé (PhD) ont été déterminés chez FMRP et par la suite extrapolés aux deux homologues. B. Distribution des FXRP chez la souris adulte. Les niveaux les plus élevés de FMRP se retrouvent dans le testicule, le cerveau et le cervelet. En ordre décroissant, on retrouve FMRP dans tous les tissus étudiés exceptés les muscles. FXR1P suit grosso modo la même tendance ; en revanche, de hauts niveaux d’isoformes particulières se retrouvent dans les muscles striés. FXR2P ne semble toutefois pas suivre les mêmes distributions que ses homologues. Les poids moléculaires apparents (kDa) sont donnés vis-à-vis de chaque protéine.
FMR1 est exprimé très tôt au cours du développement de l’embryon humain et de la souris ; son expression va en diminuant et devient par la suite plus localisée dans certains tissus. Chez l’adulte, FMRP est détectée dans presque tous les tissus, quoique à des degrés très différents (Figure 1B), avec une expression préférentielle dans le cerveau et les testicules [6]. Dans le cerveau, FMRP est très abondante dans les neurones corticaux et dans les cellules de Purkinje de la couche granulaire du cervelet. Dans le testicule, FMRP est principalement présente dans les spermatogonies. Chez la souris Fmr1-/-, des troubles d’apprentissage et du comportement, ainsi que la macroorchidie sont observés, rappelant les traits caractéristiques des patients atteints du syndrome X fragile [7]. Cela en fait un puissant outil pour l’étude des fonctions altérées à la suite de l’absence de FMRP.
La répartition tissulaire et les degrés d’expression de FXR1P suivent ceux de FMRP. Les muscles striés cardiaque et squelettiques constituent une exception, puisque seule FXR1P est exprimée sous une forme longue particulière de 82-84 kDa [8]. Les souris Fxr1-/- meurent quelques minutes après leur naissance, vraisemblablement à cause d’un développement anormal des muscles cardiaque et respiratoires [9]. En effet, FXR1P est nécessaire au développement des muscles comme nous l’avons montré récemment chez la grenouille [10]. FXR2P apparaît comme le parent négligé de la famille : peu d’études lui sont consacrées. Pourtant, la souris Fxr2-/- montre certains traits de comportement semblables à ceux de la souris Fmr1-/- [11] suggérant une certaine redondance fonctionnelle des protéines.
La famille des FXRP a été étonnamment conservée au cours de l’évolution. Tandis que les mammifères et le poisson zèbre possèdent les trois membres de la famille FXRP, la grenouille n’en compte que deux (xfmrp et xfxr1p) et la mouche qu’un seul (dfmrp). Même le cnidaire Hydratinia possède un orthologue du gène FMR1 qui est exprimé dans les cellules dites nerveuses [12]. Les cnidaires représentant le phylum métazoaire le plus ancien possédant un système nerveux, la présence d’un orthologue suggère que ces protéines ont été conservées afin d’accomplir une fonction essentielle propre au système nerveux.
FMRP et la régulation de la traduction dans les cellules « roturières »
Les premières études de caractérisation de FMRP ont été réalisées dans des lignées fibroblastiques, épithéliales et lymphoblastiques, que l’on peut qualifier de « roturières », mais ces lignées ne sont pas forcément adaptées à l’étude des fonctions d’une protéine jouant un rôle dans le développement des cellules hautement spécialisées que sont les neurones. Dans toutes les lignées utilisées, la majorité de FMRP se retrouve dans les complexes ribonucléoprotéiques (RNP) associés aux polyribosomes dits en traduction active [13]. Dans des lignées lymphoblastiques dérivées de cellules de patients X fragile, l’absence de FMRP va de pair avec une distribution altérée de certains ARNm le long des polyribosomes, confirmant le rôle essentiel joué par FMRP aux fins d’une traduction adéquate de certains ARNm [14]. Par ailleurs, diverses approches expérimentales montrent que FMRP peut se comporter comme un répresseur de la traduction. Notamment, l’expression élevée de FMRP, après transfection transitoire de cellules Fmr1-/-, induit la formation de granules dans lesquels sont emprisonnés des ARNm maintenus dans un état réprimé [15].
Comment concilier ainsi le fait que FMRP se comporte en répresseur tout en étant présente sur les polyribosomes en traduction active ? Un scénario s’échafaude à la lumière de nouvelles découvertes dans le domaine de la traduction. Des études récentes montrent que certains ARNm, tel nanos chez la mouche, peuvent être présents sur des polyribosomes tout en demeurant inactifs. L’interaction de FMRP avec la voie de l’interférence par l’ARN qui régule le clivage, la dégradation et l’inhibition de la traduction de certains ARNm, pourrait expliquer ce paradoxe. Il a été rapporté que FMRP est associée au complexe RISC, intervenant dans le métabolisme d’une nouvelle classe d’ARN non traduits, les micro-ARN (ARNmi). Ceux-ci sont clivés pour produire des ARN interférents (ARNi) s’appariant aux ARNm pour les réprimer, permettant ainsi d’exercer un contrôle local, rapide et réversible de la traduction de ces ARNm [16].
FMRP et la régulation de la traduction dans les cellules « nobles »
Bien que FMRP soit particulièrement enrichie dans les neurones, cellules « nobles » par excellence, son association aux RNP polyribosomiques dans ces cellules n’avait pas été étudiée, aussi étonnant que cela puisse paraître. Cette association n’a été validée expérimentalement que récemment [17, 18].
Chez les souris Fmr1-/-, l’absence de FMRP conduit à une répartition anormale de certains ARNm le long des polyribosomes du cerveau [14]. Plusieurs approches in vitro ont été tentées pour sélectionner ces ARNm-cibles. Deux classes d’ARNm se dessinent, avec comme motif apparent de reconnaissance soit une structure en G-quartet, soit une séquence polyU [19]. Plus récemment, un ARN synthétique possédant une structure tridimensionnelle appelée kissing complex s’est avéré capable de lier le domaine KH2 [19]. Toutefois, nous ignorons si cette structure existe dans la nature. Bien que tous ces ARNm se lient avec une forte affinité à FMRP in vitro, cette liaison ne reflète pas forcément la situation in vivo, infiniment plus complexe. Miyashiro et ses collaborateurs [20] ont identifié des cibles in vivo et montré que certains de ces ARNm subissent des changements subtils dans leur localisation et leur abondance relative chez les souris Fmr1-/-. Actuellement, toutes classes confondues, près de mille ARNm sembleraient ainsi être affectés.
FMRP comme répresseur des ARNm des granules neuronaux
Exprimée transitoirement dans des cultures primaires de neurones, une protéine chimérique FMRP-GFP s’accumule préférentiellement dans les RNPm du corps cellulaire, en association avec l’appareil de traduction. De plus, en accord avec des travaux récents [21], une population mineure et distincte de RNPm, contenant FMRP, est détectable sous forme de granules dendritiques (Figure 2A). Les trois membres de la famille FXRP se retrouvent aussi dans ces granules, qui forment des unités mobiles contenant des ARNm réprimés adressés à des destinations particulières [22]. FMRP jouerait donc un rôle dans le transport mais également dans la répression de cargos d’ARNm neuronaux.
Figure 2
Les trois membres de la famille FXRP sont co-localisés dans les granules présents dans les neurites.
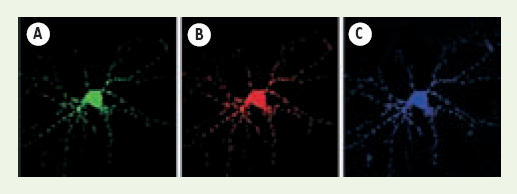
(A) FMRP avec une étiquette GFP (vert), (B) FXR1P avec une RFP (rouge) ainsi que (C) FXR2P avec une CFP (bleu) ont été exprimées après triple transfection de neurones d’hippocampe de rat en culture primaire.
Dans cet ordre d’idée, nous avons montré que FMRP se comporte comme un chaperon dont la liaison favorise une conformation stable de l’ARN. Ainsi, une ou un petit nombre de molécules de FMRP ouvrirait la structure de l’ARNm, favorisant l’amorce de sa traduction tandis qu’un plus grand nombre de FMRP, de concert avec d’autres protéines liant l’ARN, contribuerait à l'empaquetage de l’ARNm, le rendant inaccessible à l’appareil de traduction [23].
FMRP est nécessaire pour la synthèse protéique synaptique locale
Peu après la découverte de l’association de FMRP avec l’appareil de traduction [24], Weiler et ses collaborateurs [25] ont observé qu’en réponse à la stimulation des récepteurs métabotropiques au glutamate, l’ARNm de Fmr1 est rapidement recruté sur les polyribosomes pour y être traduit localement dans la synapse. Cette synthèse locale est un phénomène essentiel pour la plasticité neuronale et repose sur la présence d’ARNm apriori réprimés. La plasticité synaptique intervenant dans le développement et la maturation synaptique, sa dérégulation pourrait être à la source des altérations des fonctions cognitives supérieures observées chez les patients [26]. De plus, l’absence de FMRP dans les neurones des patients X fragile, tout comme chez la souris Fmr1-/-, se traduit par la formation d’épines dendritiques surnuméraires et immatures, laissant supposer que FMRP régule la plasticité synaptique en participant aux modifications biochimiques découlant de la synthèse locale de protéines. Cela pourrait engendrer un remodelage du cytosquelette, donc de la synapse [27].
Des p’tits granules, des p’tits granules, encor’ des p’tits granules…
Il est établi depuis plus de 30 ans qu’à la suite d’un stress imposé à la cellule, l’appareil de traduction se déstructure et les polyribosomes sont convertis en monosomes tandis que l’ARNm est relargué et, éventuellement, détruit. Ce n’est que récemment que le devenir de ces ARNm, libérés sous forme de RNPm, a été élucidé. Ces ARNm sont remisés dans des granules pour y être transitoirement réprimés et protégés, en attendant que la cellule retrouve de meilleures conditions physiologiques [28]. Ainsi, de nombreuses protéines liant l’ARNm, y compris les FXRP, peuvent être détectées dans ces granules (Figure 3A). Contre toute attente, nous avons retrouvé ce phénomène de formation de granules dans des cellules surexprimant FMRP (Figure 3B). Dès qu’un certain seuil de surcharge de FMRP est atteint, la protéine piège les ARNm et les réprime en les remisant dans des granules [15], jouant un rôle analogue à celui envisagé pour les granules neuronaux (Figure 3C). Dans le muscle, FXR1P se retrouve aussi dans des structures granulaires semblables aux costamères [10] contenant des ARNm nécessaires à la synthèse locale de protéines sollicitées dans la contraction, la réparation et l’entretien du muscle. FXR1P pourrait ainsi participer à la régulation de la traduction de ces ARNm.
Figure 3
FMRP se retrouve dans des granules (A) induits par de hauts niveaux de FMRP, (B) induits par choc thermique, (C) normalement observés dans un axone.
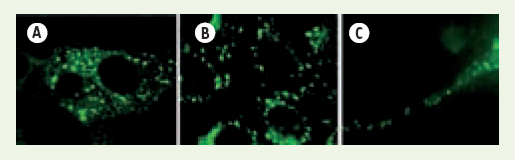
Le dénominateur commun est la formation de granules contenant l’appareil de traduction sous une forme réprimée.
Ces granules d’origines diverses partagent certains composants protéiques et il existe une interconversion possible entre les structures induites par un stress ou par la surexpression de FMRP [15] et les granules de cellules germinales nécessaires pour emmagasiner les ARNm, ainsi que les granules de transport neuronaux [29].
Conclusions
FMRP agirait comme un chaperon de l’ARNm, un répresseur de la traduction et un transporteur. La présence de FMRP dans les neurones serait donc nécessaire pour maintenir les ARNm réprimés dans les granules dendritiques et contrôler leur traduction locale dans la synapse (Figure 4).
Figure 4
Modèle illustrant le rôle de FMRP et des FXRP dans la répression de l’ARNm présent dans les granules neuronaux en route vers des destinations lointaines, telles les synapses, lieu d’une traduction hautement spécialisée.
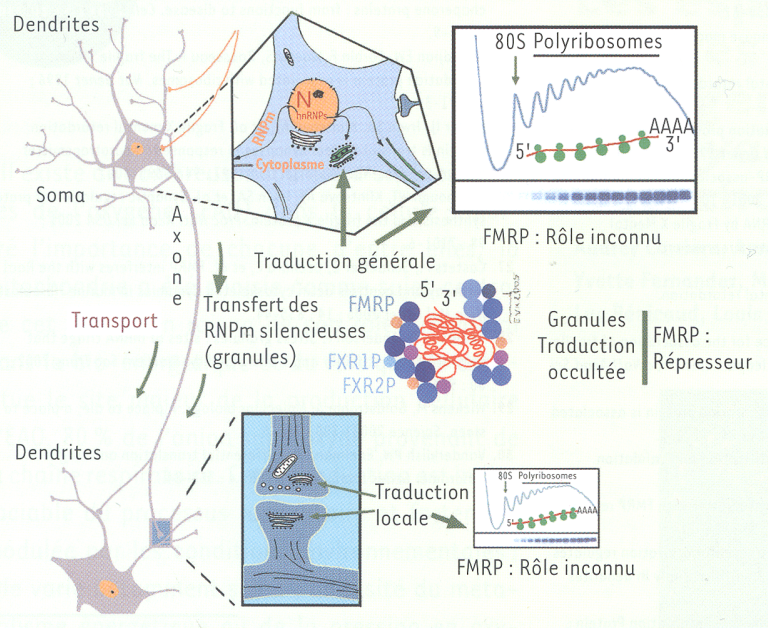
FMRP se retrouve assise sur des polyribosomes dans le corps cellulaire (soma), ainsi qu’au niveau des synapses. Les granules sont représentés par une structure compacte contenant l’ARNm, ainsi que plusieurs molécules de FMRP, FXR1P et FXR2P (en bleu) qui participeraient aux modifications conformationnelles de l’ARNm. D’autres protéines associées aux granules sont présentées en d’autres couleurs (à titre d’exemple, PABP et CPEB).
Dans un contexte plus général, il est admis que les trois membres de la famille FXRP puissent agir conjointement au sein des mêmes complexes RNPm. Cependant, la hiérarchie fonctionnelle des FXRP, ainsi que leur stoechiométrie respective ne sont pas connues. Nous proposons que dans les cellules dites « roturières », exemptes de prolongements cellulaires, FXR1P et FXR2P peuvent compenser l’absence de FMRP. En revanche, dans les neurones, cette absence pourrait avoir comme conséquence une répression incomplète de certains ARNm qui seraient alors traduits inadéquatement en lieu et en temps, conduisant à une altération du développement et de la connexité synaptique. Des altérations dans la topologie synaptique du cerveau contribueraient ainsi à expliquer le retard mental observé chez les patients X fragile, sans toutefois éclaircir la grande diversité des phénotypes observés.
Depuis la découverte du gène FMR1 en 1991, le nombre de laboratoires qui se joignent aux recherches sur les fonctions de FMRP ne cesse d’augmenter. Cette croissance illustre l’intérêt que FMRP suscite dans diverses sphères de la recherche en raison de son importance chez l’humain à la fois dans le développement du système nerveux et dans celui des fonctions cognitives supérieures. D’où l’intérêt récent [30] pour FMRP de Gerald Edelman, concepteur de la théorie de la Sélection des Groupes Neuronaux (Theory of Neuronal Group Selection), d’après laquelle des populations de neurones développent des réseaux interconnectés selon une sélection darwinienne.
Parties annexes
Références
- 1. Bardoni B, Mandel JL Advances in understanding of fragile X pathogenesis and FMRP function, and in identification of X linked mental retardation genes. Curr Opin Genet Dev 2002 ; 12 : 284-93.
- 2. O’Donnell WT, Warren ST. A decade of molecular studies of fragile X syndrome. Annu Rev Neurosci 2002 ; 25 : 315-38.
- 3. Hagerman PJ, Cronister A. Fragile X syndrome : diagnosis, treatment and research. Baltimore : University Press, 1996.
- 4. Khandjian EW. Biology of the fragile X mental retardation protein, an RNA-binding protein. Biochem Cell Biol 1999 ; 77 : 331-42.
- 5. Mazroui R, Huot ME, Tremblay S, et al. Fragile X Mental Retardation protein determinants required for its association with polyribosomal mRNPs. Hum Mol Genet 2003 ; 12 : 3087-96.
- 6. Devys D, Lutz Y, Rouyer N, et al. The FMR-1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nat Genet 1993 ; 4 : 335-40.
- 7. Consortium TD-BFX. Fmr1 knockout mice : a model to study fragile X mental retardation. The Dutch-Belgian Fragile X Consortium. Cell 1994 ; 78 : 23-33.
- 8. Khandjian EW, Bardoni B, Corbin F, et al. Novel isoforms of the fragile X related protein FXR1P are expressed during myogenesis. Hum Mol Genet 1998 ; 7 : 2121-8.
- 9. Mientjes EJ, Willemsen R, Kirkpatrick LL, et al. Fxr1 knockout mice show a striated muscle phenotype : implications for Fxr1p function in vivo. Hum Mol Genet 2004 ; 13 : 1291-302.
- 10. Huot ME, Bisson N, Davidovic L, et al. The RNA-binding protein Fragile X-related 1 regulates somite formation in Xenopus laevis. Mol Biol Cell 2005 ; 16 : 4350-61.
- 11. Bontekoe CJ, McIlwain KL, Nieuwenhuizen IM, et al. Knockout mouse model for Fxr2 : a model for mental retardation. Hum Mol Genet 2002 ; 11 : 487-98.
- 12. Guduric-Fuchs J, Mohrlen F, Frohme M, Frank U. A fragile X mental retardation-like gene in a cnidarian. Gene 2004 ; 343 : 231-8.
- 13. Corbin F, Bouillon M, Fortin A, et al. The fragile X mental retardation protein is associated with poly(A)+ mRNA in actively translating polyribosomes. Hum Mol Genet 1997 ; 6 : 1465-72.
- 14. Brown V, Jin P, Ceman S, et al. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell 2001 ; 107 : 477-87.
- 15. Mazroui R, Huot ME, Tremblay S, et al. Trapping of messenger RNA by Fragile X Mental Retardation protein into cytoplasmic granules induces translation repression. Hum Mol Genet 2002 ; 11 : 3007-17.
- 16. Jin P, Alisch RS, Warren ST. RNA and microRNAs in fragile X mental retardation. Nat Cell Biol 2004 ; 6 : 1048-53.
- 17. Khandjian EW, Huot ME, Tremblay S, et al. Biochemical evidence for the association of fragile X mental retardation protein with brain polyribosomal ribonucleoparticles. Proc Natl Acad Sci USA 2004 ; 101 : 13357-62.
- 18. Stefani G, Fraser CE, Darnell JC, Darnell RB. Fragile X mental retardation protein is associated with translating polyribosomes in neuronal cells. J Neurosci 2004 ; 24 : 7272-6.
- 19. Darnell JC, Mostovetsky O, Darnell RB. FMRP RNA targets : identification and validation. Genes Brain Behav 2005 ; 4 : 341-9.
- 20. Miyashiro KY, Beckel-Mitchener A, Purk TP, et al. RNA cargoes associating with FMRP reveal deficits in cellular functioning in Fmr1 null mice. Neuron 2003 ; 37 : 417-31.
- 21. Antar LN, Afroz R, Dictenberg JB, et al. Metabotropic glutamate receptor activation regulates fragile X mental retardation protein and FMR1 mRNA localization differentially in dendrites and at synapses. J Neurosci 2004 ; 24 : 2648-55.
- 22. Davidovic L, Tremblay S, De Koninck P, Khandjian EW. Fragile X Mental Retardation Protein : To be or not to be a Translational Repressor. In : Sung YJ, Denman RB, eds. The molecular basis of fragile X syndrome. Kerala (India) : Research Signpost, 2005 : 129-43.
- 23. Ivanyi-Nagy R, Davidovic L, Khandjian EW, Darlix JL. Disordered RNA chaperone proteins : from functions to disease. Cell Mol Life Sci 2005 ; 62 : 1-9.
- 24. Khandjian EW, Corbin F, Woerly S, Rousseau F. The fragile X mental retardation protein is associated with ribosomes. Nat Genet 1996 ; 12 : 91-3.
- 25. Weiler IJ, Irwin SA, Klintsova AY, et al. Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc Natl Acad Sci USA 1997 ; 94 : 5395-400.
- 26. Greenough WT, Klintsova AY, Irwin SA, et al. Synaptic regulation of protein synthesis and the fragile X protein. Proc Natl Acad Sci USA 2001 ; 98 : 7101-6.
- 27. Castets M, Schaeffer C, Bechara E, et al. FMRP interferes with the Rac1 pathway and controls actin cytoskeleton dynamics in murine fibroblasts. Hum Mol Genet 2005 ; 14 : 835-44.
- 28. Kedersha N, Anderson P. Stress granules : sites of mRNA triage that regulate mRNA stability and translatability. Biochem Soc Trans 2002 ; 30 : 963-9.
- 29. Wickens M, Goldstrohm A. Molecular biology. A place to die, a place to sleep. Science 2003 ; 300 : 753-5.
- 30. Vanderklish PW, Edelman GM. Differential translation and fragile X syndrome. Genes Brain Behav 2005 ; 4 : 360-84.
Liste des figures
Figure 1
Structure des membres de la famille FXRP et distribution de ces protéines dans divers tissus et organes de la souris adulte.
A. Structure des protéines FXR : les barres verticales (en vert foncé) correspondent aux acides aminés (aa) identiques, tandis que les barres horizontales (en rouge) représentent les aa divergents. Les domaines fonctionnels correspondant aux signaux de localisation nucléaire (NLS) et d’export nucléaire (NES), aux motifs de reconnaissance de l’ARN (KH1, KH2 et RGG), au domaine d’interaction protéine-protéine (PPID) et enfin au domaine phosphorylé (PhD) ont été déterminés chez FMRP et par la suite extrapolés aux deux homologues. B. Distribution des FXRP chez la souris adulte. Les niveaux les plus élevés de FMRP se retrouvent dans le testicule, le cerveau et le cervelet. En ordre décroissant, on retrouve FMRP dans tous les tissus étudiés exceptés les muscles. FXR1P suit grosso modo la même tendance ; en revanche, de hauts niveaux d’isoformes particulières se retrouvent dans les muscles striés. FXR2P ne semble toutefois pas suivre les mêmes distributions que ses homologues. Les poids moléculaires apparents (kDa) sont donnés vis-à-vis de chaque protéine.
Figure 2
Les trois membres de la famille FXRP sont co-localisés dans les granules présents dans les neurites.
(A) FMRP avec une étiquette GFP (vert), (B) FXR1P avec une RFP (rouge) ainsi que (C) FXR2P avec une CFP (bleu) ont été exprimées après triple transfection de neurones d’hippocampe de rat en culture primaire.
Figure 3
FMRP se retrouve dans des granules (A) induits par de hauts niveaux de FMRP, (B) induits par choc thermique, (C) normalement observés dans un axone.
Le dénominateur commun est la formation de granules contenant l’appareil de traduction sous une forme réprimée.
Figure 4
Modèle illustrant le rôle de FMRP et des FXRP dans la répression de l’ARNm présent dans les granules neuronaux en route vers des destinations lointaines, telles les synapses, lieu d’une traduction hautement spécialisée.
FMRP se retrouve assise sur des polyribosomes dans le corps cellulaire (soma), ainsi qu’au niveau des synapses. Les granules sont représentés par une structure compacte contenant l’ARNm, ainsi que plusieurs molécules de FMRP, FXR1P et FXR2P (en bleu) qui participeraient aux modifications conformationnelles de l’ARNm. D’autres protéines associées aux granules sont présentées en d’autres couleurs (à titre d’exemple, PABP et CPEB).