Abstracts
Résumé
Les maladies héréditaires du métabolisme ou erreurs innées du métabolisme sont des maladies rares, mais très diverses, puisqu’on estime leur nombre à 500. Ce chiffre explique les difficultés rencontrées dans les diagnostics et les traitements. Ces maladies sont dues à des mutations de gènes codant pour des protéines impliquées dans les voies métaboliques. Les maladies héréditaires du métabolisme les plus fréquentes sont dues à l’accumulation de certains intermédiaires ou métabolites, ou correspondent à des déficits énergétiques, par exemple les déficits de la chaîne respiratoire, de l’oxydation des acides gras ou du métabolisme glucidique, ou encore à des perturbations du métabolisme de molécules complexes. Les biochimistes apportent une aide souvent essentielle au diagnostic et au suivi du traitement nutritionnel ou médicamenteux. Les diagnostics biochimiques reposent principalement sur des dosages de métabolites et des mesures d’activités enzymatiques, plus rarement sur la recherche de mutations. Une meilleure prise en charge des patients concernés impose une amélioration du dépistage néonatal et l’accroissement de l’efficacité de laboratoires de biochimie spécialisée. C’est pour cela que se développe la métabolomique, qui regroupe l’ensemble des approches technologiques permettant de doser un très grand nombre de métabolites. Parmi ces approches, la spectrométrie de masse en tandem constitue une méthode de choix.
Summary
Both hope and illusion, the recent progress in biology has raised our expectations that one day it will be possible to introduce a biological sample into an apparatus which will then deliver in a few minutes thousands of qualitative and quantitative data concerning the genome, transcriptome, proteome and metabolome, thereby contributing to diagnosis and follow-up of diseases which are now difficult to identify. Such machines do not exist yet and, in any case, should be associated with the appropriate and adequate clinical work on the disease and with the patient. This « total » approach is of course being pushed by the recent decoding of the genomes of several species (genomics), the development of high throughput analysis of mRNAs (transcriptomics), and the efforts to identify the protein products on a large scale (proteomics). Wide sectors of medicine are waiting for the results of these new medium and high throughput technological approaches, for example, in order to identify early markers of diseases. The object of this article is to present a biochemist’s point of view on hereditary metabolic diseases (also referred to as inborn errors of metabolism), a field of medicine and research covering very diverse clinical and biochemical aspects. Significant advances will be made possible by improving the present methods of analysis of the metabolome which establishes a link between genotypes and phenotypes, an area now called metabolomics. The contribution of proteomics will be important as well but will still require some time.
Article body
À la fois espoir et illusion, les progrès récents en biologie laissent penser qu’il sera un jour possible d’introduire un échantillon biologique dans un appareil qui livrera en quelques minutes des milliers de données qualitatives et quantitatives concernant le génome, transcriptome, protéome et métabolome, aidant ainsi au diagnostic et au suivi de maladies difficiles à identifier. De tels appareils n’existent pas (encore) et ne devront, en tout cas, jamais être utilisés séparément d’une approche clinique adéquate. Cette approche « totale » est évidemment stimulée par le déchiffrage récent du génome de plusieurs espèces, la génomique, et le développement de l’analyse à haut débit des ARN messagers, la transcriptomique. À cela s’ajoutent les efforts d’identification à grande échelle des produits protéiques, ou protéomique. De larges secteurs de la médecine attendent des retombées de ces approches technologiques nouvelles à moyen ou haut débit, par exemple l’identification de marqueurs précoces des maladies. Cet article, qui représente le point de vue du biochimiste, a pour objet de présenter les maladies héréditaires du métabolisme, un secteur de la médecine et de la recherche couvrant des aspects cliniques et biochimiques très divers, qui ne pourra progresser que via l’amélioration des méthodes analytiques de ce qui est maintenant nommé la métabolomique. L’analyse du métabolome établit un lien entre les génotypes et les phénotypes biologiques. L’apport de la protéomique sera également important, mais demandera encore du temps.
Maladies héréditaires du métabolisme
Il y a environ 500 maladies métaboliques héréditaires, nommées aussi erreurs innées du métabolisme [1-5]. Ces maladies illustrent la diversité et la complexité des voies métaboliques. Elles sont dues à des mutations de gènes qui altèrent l’activité de protéines (enzymes, transporteurs, protéines de structure), ce qui se traduit par des taux anormaux (diminution, absence, accumulation…) de métabolites et l’apparition de signes cliniques. Ces maladies sont rares et souvent graves, chacune concernant moins d’une naissance sur 10 000. Le nombre de ces maladies pouvant être traitées était faible, mais il s’accroît régulièrement, la qualité du traitement dépendant étroitement de la rapidité du diagnostic et d’une coopération solide entre cliniciens experts et biochimistes spécialisés. Dans la plupart des cas, les traitements sont diététiques, et parfois médicamenteux. Pour les cliniciens et les biochimistes, ces maladies se présentent sur un mode soit aigu, soit chronique.
Ces maladies sont classées en trois grands groupes : (1) les maladies dues à une intoxication endogène : aminoacidopathies, aciduries organiques, déficits du cycle de l’urée, anomalies du métabolisme du galactose, maladies des métaux ; (2) les maladies du métabolisme énergétique : déficits de la chaîne respiratoire, de l’oxydation des acides gras, de la néoglucogenèse, du métabolisme des corps cétoniques, glycogénoses, hypoglycémies induites par hyper-insulinisme ; (3) les maladies du métabolisme des molécules complexes : erreurs de la synthèse du cholestérol, du métabolisme des polyols, de la glycosylation des protéines, maladies des peroxysomes, des lysosomes…
Classification biochimique des maladies métaboliques
Du point de vue du biochimiste, on peut classer les maladies métaboliques en deux grandes catégories. D’une part, les maladies du métabolisme intermédiaire, par exemple du métabolisme de certains acides aminés ou de l’oxydation des acides gras, d’autre part, les maladies du métabolisme de molécules complexes impliquant, par exemple, les leucotriènes, les polyols, les polysaccharides, les glycosylations.
Maladies du métabolisme intermédiaire
Maladies du métabolisme des acides aminés
Les aminoacidopathies correspondent à des anomalies de la dégradation des acides aminés. Lorsque le déficit implique une enzyme située au début de la voie de dégradation, l’aminoacidopathie se traduit par une accumulation d’acides aminés. Les exemples sont la phénylcétonurie et la leucinose. Lorsque le déficit implique une enzyme distale dans la voie de dégradation, il peut y avoir accumulation d’acides aminés, mais il y a aussi accumulation d’acides organiques produits à partir de dérivés du coenzyme A. On parle alors d’acidémies organiques, ou d’aciduries organiques (aciduries propionique, méthylmalonique, glutarique…), car elles sont souvent diagnostiquées à partir de dosages urinaires. La dégradation des acides aminés produisant de l’ammonium, une substance neurotoxique normalement convertie en urée et excrétée dans l’urine, des déficits en enzymes du cycle de l’urée impliquées dans la détoxification de l’ammonium peuvent induire une hyperammoniémie dangereuse pour le cerveau.
Maladies de l’oxydation des acides gras et de la cétogenèse
Lors du jeûne, l’énergie provient principalement de l’oxydation mitochondriale totale des acides gras ou de leur oxydation partielle sous forme de corps cétoniques dans le foie. Des déficits enzymatiques de ces voies d’oxydation des acides gras induisent une hypoglycémie hypocétosique et des troubles aggravés par le jeûne. Le diagnostic se fait par les dosages des acides gras libres et des corps cétoniques plasmatiques, le dosage des acides organiques urinaires, l’analyse du profil des acylcarnitines. Ces patients doivent absolument éviter de jeûner.
Maladies du métabolisme des sucres et de leur transport
Il s’agit de situations diverses impliquant le métabolisme du galactose, du fructose, du glycérol, les voies de la néoglucogenèse, celles de la synthèse ou l’hydrolyse du glycogène, les mécanismes de transport du glucose. Les situations de galactosémie ou de fructosémie induisent l’accumulation de métabolites pathogènes et impliquent l’exclusion du galactose et du fructose alimentaires. Des déficits touchant la voie de la néoglucogenèse se traduisent par une hypoglycémie et une acidémie lactique lors de courtes périodes de jeûne. Les glycogénoses sont multiples. Par exemple, la glycogénose de type Ia est due à un déficit en glucose-6 phosphatase.
Erreurs de biosynthèse ou de dégradation des molécules complexes
Maladies peroxysomales
Les rôles biochimiques des peroxysomes sont très divers et incluent, par exemple, la biosynthèse du cholestérol. Des erreurs dans la biogenèse des peroxysomes ou l’oxydation des acides gras à longue chaîne, ou dans le métabolisme de l’acide phytanique, induisent des états pathologiques graves tels que le syndrome de Zellweger ou la maladie de Refsum. Une autre maladie peroxisomale d’intérêt impliquant le défaut d’oxydation des acides gras à très longue chaîne est l’adrénoleucodystrophie liée à l’X. Cette maladie est due à un déficit du transporteur d’acides gras à très longue chaîne [6]. D’autres syndromes résultent de déficits dans la biosynthèse des éthers de phospholipides, de déficit en catalase ou encore d’erreur dans le métabolisme du glyoxylate.
Maladies de surcharge lysosomale
Des déficits d’hydrolases de mucopolysaccharides ou de sphingolipides provoquent des accumulations de substrats. La maladie de Fabry est due à un déficit en α-glucosidase, la maladie de Gaucher résulte d’un déficit en glucocérébrosidase. La cystinose est causée par un déficit de transport lysosomal.
Maladies métaboliques mitochondriales
Il s’agit de maladies impliquant la pyruvate-déshydrogénase, les enzymes du cycle de Krebs ou les complexes de la chaîne respiratoire. Les problèmes d’oxydation des acides gras ont été mentionnés ci-dessus. Fréquemment, une acidose lactique est observée, accompagnant des troubles neuromusculaires. Des mutations dans les génomes nucléaires et mitochondriaux se partagent la responsabilité de ces maladies. Le degré d’hétéroplasmie[1] mitochondriale contribue à la diversité des phénotypes observés parmi les patients porteurs d’une modification de l’ADN mitochondrial.
Maladies des purines et pyrimidines
Il s’agit de maladies diverses provoquées par des déficits enzymatiques de la synthèse ou de la dégradation des purines ou des pyrimidines. Ainsi, une maladie grave du système immunitaire causée par le déficit en adénosine-désaminase, l’immunodéficience combinée sévère, ou une maladie neuromusculaire sévère due au déficit en hypoxanthine-guanine phosphoribosyltranférase, la maladie de Lesch-Nyhan.
Erreurs de glycosylation des protéines
De nombreuses enzymes, environ une trentaine, contrôlent les phénomènes de glycosylation. Des déficits d’activité de ces enzymes sont à l’origine de maladies rassemblées sous le terme de syndromes CDG (congenital disorders of glycosylation).
De nombreuses autres maladies héréditaires du métabolisme ne seront que citées : les maladies du métabolisme de la cobalamine (vitamine B12) et des folates, qui induisent une acidurie méthylmalonique, celles concernant le transport intestinal ou rénal de certains acides aminés, celles du métabolisme du cuivre, du fer ou encore du zinc, les maladies de la synthèse des isoprénoïdes ou des stérols, les erreurs de synthèse des acides biliaires, de synthèse de l’hème (porphyries), les maladies du métabolisme des lipoprotéines, les déficits de synthèse des neurotransmetteurs…
Méthodes actuelles de diagnostic biochimique et traitement des déficits métaboliques
Le diagnostic précis et le suivi des maladies héréditaires du métabolisme repose, d’une part, sur des examens classiques d’orientation (glycémie, cétose, ammoniémie…) suivis par l’utilisation de méthodes d’analyse biochimique complexes telles que la chromatographie des acides aminés ou la chromatographie en phase gazeuse couplée à la spectrométrie de masse des acides organiques, l’analyse des acylcarnitines, des dosages enzymatiques particuliers (certains étant faits dans le cadre de recherche de diagnostic prénatal), et la recherche de mutations de l’ADN. Si les dosages de métabolites ou la mesure d’activités enzymatiques, qui fournissent des données phénotypiques, sont généralement préférées aux recherches de mutations, cette approche est cependant utile à la recherche ou la confirmation de plusieurs maladies héréditaires du métabolisme. Les analyses d’ADN sont évidemment importantes pour les études de transmission familiale, le diagnostic prénatal et le conseil génétique (Figure 1).
Figure 1
Organisation du diagnostic des maladies héréditaires du métabolisme dans un laboratoire de biochimie spécialisée.
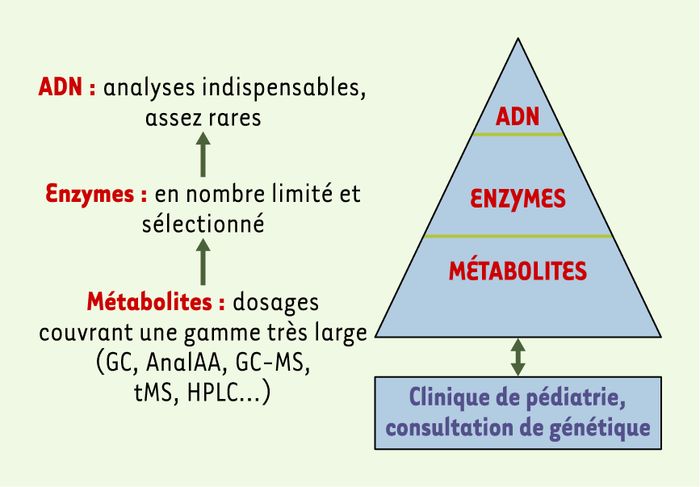
Les activités du laboratoire de biochimie sont représentées par un triangle. Pour contribuer au diagnostic biochimique et au suivi des traitements, le laboratoire effectue des analyses de nombreux métabolites (GC : chromatographie en phase gazeuse ; AnalAA : analyse d’acides aminés ; MS : spectrométrie de masse ; tMS : spectrométrie de masse en tandem ; HPLC : chromatographie liquide haute performance). Les analyses de métabolites constituent le principal de l’activité. Cependant, l’information essentielle peut venir de dosages enzymatiques ou de l’identification de mutations.
Certaines maladies héréditaires du métabolisme peuvent être traitées via une prise en charge diététique adaptée. Les aminoacidopathies et certaines aciduries organiques sont traitées par restriction alimentaire en protéines ou en certains acides aminés. Certaines maladies sont traitées par apport de vitamines (par exemple, la thiamine), de cofacteurs tels que le coenzyme Q, ou de carnitine, dans le cas du rare déficit héréditaire de capture cellulaire de la carnitine. Des voies artificielles d’épuration de l’azote peuvent être créées par administration de benzoate. D’autres stratégies thérapeutiques plus sophistiquées peuvent consister à inhiber spécifiquement une étape en amont du déficit enzymatique de manière à privilégier l’accumulation d’une substance moins dangereuse, ou à régénérer une voie métabolique défaillante en fournissant les molécules carbonées adéquates (anaplérose[2] du cycle de Krebs, par exemple). Dans de très rares situations (déficit en adénosine-désaminase), des tentatives de thérapie génique ont débuté [7]. Par ailleurs, l’enzymothérapie substitutive est utilisée pour traiter quelques maladies dont certaines maladies de surcharge des lysosomes [8].
Les bénéfices de la protéomique dans l’étude des maladies métaboliques sont encore discrets. Les attentes sont doubles : d’une part, l’identification de marqueurs utiles au diagnostic et au suivi des traitements, d’autre part, la compréhension des mécanismes conduisant à la situation pathologique. En effet, dans plusieurs maladies métaboliques héréditaires, si l’enzyme défectueuse est identifiée, les mécanismes responsables du phénotype restent mystérieux. Dans le domaine de l’identification et du dosage de métabolites, plusieurs méthodes sont en plein développement.
Développement de la métabolomique
La très grande diversité des maladies héréditaires du métabolisme et des activités enzymatiques défectueuses doit être comparée à l’efficacité relativement limitée des méthodes d’analyse généralement utilisées. Cette situation explique que, même si de nombreux diagnostics biochimiques sont effectués par des centres spécialisés, trop peu de maladies sont expliquées sur le plan biochimique. De plus, la possibilité de soigner efficacement les patients exige des diagnostics rapides, souvent dans les premiers jours suivant la naissance. La tâche est donc immense et les progrès ne peuvent venir que d’une évolution technologique importante.
Cette évolution concerne principalement ce que l’on désigne sous le néologisme de « métabolomique », c’est-à-dire un ensemble de technologies permettant d’identifier et doser un très grand nombre de métabolites, témoins de l’activité des milliers d’enzymes dont le déficit est responsable des maladies héréditaires du métabolisme. Ces technologies existent déjà et correspondent à divers types de chromatographie comme l’HPLC (chromatographie liquide haute performance), la chromatographie en phase gazeuse couplée à la spectrométrie de masse, la spectrométrie de masse en tandem, la résonance magnétique nucléaire, la spectrométrie de résonance magnétique in vivo (Tableau I). En fait, le développement de la métabolomique ne consiste pas tant à mettre au point de nouveaux appareillages qu’à équiper les laboratoires de biochimie spécialisée existants avec les appareillages modernes disponibles et organiser des plates-formes technologiques. En aucun cas, il n’est question de remplacer les appareils de chromatographie en phase gazeuse couplés à un spectromètre de masse par un spectromètre de masse en tandem ; il s’agit d’amener ce second appareil dans un laboratoire disposant déjà du premier. La métabolomique permet d’identifier de nouvelles maladies héréditaires du métabolisme [9, 10]. Les méthodes de choix sont donc la spectrométrie de masse en tandem, la résonance magnétique nucléaire et la spectrométrie de résonance magnétique in vivo (MRS).
Tableau I
Quelques exemples de métabolites dosés dans le cadre de recherche de diagnostic biochimique de maladies héréditaires du métabolisme.

Cette liste ne représente qu’une petite partie des analyses nécessaires à la compréhension du métabolome du patient. IEC : chromatographie sur colonne échangeuse d’ions ; GC : chromatographie en phase gazeuse ; GC-MS : chromatographie en phase gazeuse couplée à la spectrométrie de masse ; tMS : spectrométrie de masse en tandem ; TLC : chromatographie en couche mince ; 1H-NMR : résonance magnétique nucléaire du proton ; MRS in vivo : spectrométrie de résonance magnétique in vivo.
Spectrométrie de masse en tandem
La chromatographie en phase gazeuse couplée à la spectrométrie de masse est utilisée pour analyser les acides organiques (environ 350 composés différents). Depuis plusieurs années, la spectrométrie de masse en tandem est utilisée pour l’analyse des maladies héréditaires du métabolisme. Cette technique, qui s’ajoute sans la remplacer à la chromatographie en phase gazeuse couplée à la spectrométrie de masse, est fondée sur l’utilisation de trois quadripôles permettant la production d’ions, leur séparation et leur sélection. Cette technique, rapide et précise, est en particulier utilisée avec succès pour analyser les dérivés acylcarnitines dans les maladies dues à des erreurs d’oxydation des acides gras. L’analyse des acylcarnitines permet ainsi d’essayer d’identifier un grand nombre de déficits enzymatiques distincts. Chez les nouveau-nés, cette approche permet de mettre en évidence, par exemple, le déficit en MCAD (medium-chain acylCoA dehydrogenase) qui se traduit par une accumulation d’octanoylcarnitine et de décénoylcarnitine. Le déficit en MCAD chez le nouveau-né est aussi fréquent que la phénylcétonurie (une naissance sur 15 000). La spectrométrie de masse en tandem peut aussi aider à diagnostiquer diverses aciduries organiques et être utilisée pour l’analyse des acides aminés. La spectrométrie de masse en tandem est une technique très flexible permettant d’analyser un nombre considérable de molécules.
Résonance magnétique nucléaire (RMN)
L’analyse par spectrométrie RMN des liquides biologiques (plasma, urine, liquide céphalorachidien) est une technique qui peut aussi contribuer à la compréhension des maladies héréditaires du métabolisme. En particulier, la RMN du proton peut être utilisée pour identifier et doser un très grand nombre de métabolites dont il est impossible de donner la liste ici. La RMN du phosphore et celle du carbone sont aussi utilisées. La RMN peut être la méthode de choix pour aider à comprendre une situation pathologique non élucidée. Elle peut apporter des informations structurales sur l’identité d’un métabolite inconnu repéré via une autre technique chez un patient. Elle peut être utilisée pour confirmer un diagnostic établi à partir d’une autre approche analytique. La RMN peut être requise pour la quantification de composés difficilement mesurables autrement. Récemment, elle a permis l’identification de plusieurs nouvelles maladies héréditaires du métabolisme [11].
Spectrométrie de résonance (MRS) in vivo
La recherche dans le domaine de maladies héréditaires du métabolisme bénéficie du développement récent des progrès de l’imagerie associée à la résonance magnétique (magnetic resonance imaging, MRI) et surtout de la spectrométrie de résonance magnétique in vivo (magnetic resonance spectrometry, MRS). Cette approche est en plein essor. L’analyse par MRS localisée du cerveau permet d’apprécier la situation métabolique in vivo et la détection, par exemple, de déficits en créatine (déficit en guanidinoacétate méthyltransférase), ou encore des perturbations du métabolisme des polyols ou d’un excès de N-acétyl-aspartate ou de choline. Elle est utilisée dans l’étude des encéphalopathies néonatales [12]. Récemment, la MRS a été utilisée pour doser certains neurotransmetteurs (glycine, GABA) dans le cerveau [13].
Conclusions
Des progrès dans le diagnostic des maladies héréditaires du métabolisme dépendront de l’analyse du métabolome. Sans compter les progrès faits directement par les cliniciens et les généticiens, l’amélioration du diagnostic des maladies héréditaires du métabolisme viendra d’une meilleure analyse du métabolome des patients et aussi d’une augmentation des types d’examens réalisés rapidement après la naissance. Il est fondamental que les laboratoires spécialisés puissent disposer des techniques modernes qui permettront le diagnostic et aideront à la prise en charge de ces maladies. Cela soulève d’ailleurs la question de l’amélioration du dépistage néonatal dans notre pays. Plusieurs nations ont mis en place l’utilisation de la spectrométrie de masse en tandem dans le cadre du dépistage systématique de 10 à 20 maladies héréditaires du métabolisme [14]. Une telle mise en place n’est pas sans problème. Elle suscite de nombreuses questions médicales et humaines et fait l’objet d’un débat dans plusieurs pays, dont le nôtre.
Appendices
Remerciements
L’auteur remercie les Professeurs et Docteurs Jean-Marie Saudubray, Daniel Rabier, Guy Touati, Jean-Charles Deybach et Cornelis Jakobs pour des discussions ou la lecture du manuscrit, le Dr Charlotte Sumida pour le résumé en anglais. Les activités du laboratoire de Biochimie Spécialisée de l’Hôpital Necker-Enfants Malades sont soutenues par l’Assistance Publique des Hôpitaux de Paris et la Fondation Jérôme-Lejeune.
Notes
-
[1]
L’hétéroplasmie, phénomène observé dans un nombre important de maladies héréditaires mitochondriales, correspond à la présence dans les cellules ou les tissus des malades de deux types d’ADN mitochondrial, muté et non muté.
-
[2]
L’anaplérose correspond aux réactions métaboliques permettant de maintenir le pool des intermédiaires du cycle de Krebs.
Références
- 1. Scriver CR, Beaudet AL, Sly WY, Valle D. The metabolic and molecular basis of inherited diseases. New York : MacGraw-Hill, 1995 : 2 284 p.
- 2. Fernandes J, Saudubray JM, van den Bergue G. Inborn metabolic diseases. Berlin : Springer-Verlag, 2000 : 468 p.
- 3. Hoffmann GF, Nyhan WL, Zschocke J, et al. Inherited metabolic disease. Philadelphia : Lippincott, 2002 : 436 p.
- 4. Saudubray JM, Nassogne MC, de Lonlay P, et al. Clinical approach to inherited metabolic disorders in neonates : an overview. Semin Neonatol 2002 ; 7 : 3-15.
- 5. Zschocke J, Hoffmann GF. Vademecum metabolicum. Friedrichsdorf : Milupa, 1999 : 112 p.
- 6. Aubourg P, Mandel JL. X-linked adrenoleukodystrophy. Ann NY Acad Sci 1996 ; 804 : 471-6.
- 7. Cavazzana-Calvo M, Hacein-Bey S, de Saint Basile G, et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science 2000 ; 288 : 669-72.
- 8. Winkel LP, Van den Hout JM, Kamphoven JH, et al. Enzyme replacement therapy in late-onset Pompe’s disease : a three-year follow-up. Ann Neurol 2004 ; 55 : 495-502.
- 9. Moolenaar SH, van der Knaap MS, Engelke UF, et al. In vivo and in vitro NMR spectroscopy reveal a putative novel inborn error involving polyol metabolism. NMR Biomed 2001 ; 14 : 167-76.
- 10. Huck JH, Struys EA, Verhoeven NM, et al. Profiling of pentose phosphate pathway intermediates in blood spots by tandem mass spectrometry : application to transaldolase deficiency. Clin Chem 2003 ; 49 : 1375-80.
- 11. Moolenaar SH, Engelke UFH, Hoenderop SM, et al.Handbook of 1H-NMR spectroscopy in inbornn errors of metabolism. Heilbronn : SHS International, 2002 : 82 p.
- 12. Bartha AI, Foster-Barber A, Miller SP, et al. Neonatal encephalopathy : association of cytokines with MR spectroscopy and outcome. Pediatr Res 2004 ; 56 : 960-96.
- 13. Novotny EJ Jr, Fulbright RK, Pearl PL, et al. Magnetic resonance spectroscopy of neurotransmitters in human brain. Ann Neurol 2003 ; 54 : S25-31.
- 14. Zytkovicz TH, Fitzgeralg EF, Marsden D, et al. Tandem mass spectroscopy for amino, organic and fatty acid disorders in newborn dried blood spots : a two-year summary from the New England newborn screening program. Clin Chem 2001 ; 47 : 1945-55.
List of figures
Figure 1
Organisation du diagnostic des maladies héréditaires du métabolisme dans un laboratoire de biochimie spécialisée.
Les activités du laboratoire de biochimie sont représentées par un triangle. Pour contribuer au diagnostic biochimique et au suivi des traitements, le laboratoire effectue des analyses de nombreux métabolites (GC : chromatographie en phase gazeuse ; AnalAA : analyse d’acides aminés ; MS : spectrométrie de masse ; tMS : spectrométrie de masse en tandem ; HPLC : chromatographie liquide haute performance). Les analyses de métabolites constituent le principal de l’activité. Cependant, l’information essentielle peut venir de dosages enzymatiques ou de l’identification de mutations.
List of tables
Tableau I
Quelques exemples de métabolites dosés dans le cadre de recherche de diagnostic biochimique de maladies héréditaires du métabolisme.
Cette liste ne représente qu’une petite partie des analyses nécessaires à la compréhension du métabolome du patient. IEC : chromatographie sur colonne échangeuse d’ions ; GC : chromatographie en phase gazeuse ; GC-MS : chromatographie en phase gazeuse couplée à la spectrométrie de masse ; tMS : spectrométrie de masse en tandem ; TLC : chromatographie en couche mince ; 1H-NMR : résonance magnétique nucléaire du proton ; MRS in vivo : spectrométrie de résonance magnétique in vivo.