Abstracts
Résumé
L’épidémie mondiale de syndrome métabolique - défauts des métabolismes glucidique et lipidique, obésité abdominale, dyslipidémie et hypertension, risques d’obésité, de diabète de type 2 et de maladie cardiovasculaire - reflète la diversité des influences géoclimatiques et culturelles subies par les populations concernées et le caractère soudain des changements (alimentation pléthorique et/ou déséquilibrée, sédentarisation). Outre l’héritage progressif d’un « génotype économe », accumulé au cours de siècles ponctués par les famines, les individus des générations actuelles ont subi des altérations de leur programmation épigénétique, d’une part au cours de leur développement foetal et postnatal, en liaison avec une nutrition déséquilibrée et des désordres métaboliques maternels et, d’autre part, au cours de leur vie, en liaison avec leurs excès alimentaires et l’insuffisance de leur activité physique. Afin de convertir ce « phénotype économe », aujourd’hui devenu obsolète, en un « phénotype gaspilleur », il faut identifier les séquences concernées - gènes, soumis à empreinte ou non, ou transposons, procéder au décryptage des signaux épigénétiques en cause, de leur verrouillage ou de leur labilité, puis identifier ou concevoir des molécules (nutriments et médicaments) capables de prévenir (ou de modifier) un formatage épigénétique aberrant et inadapté.
Summary
The importance of epigenetic alterations has been acknowledged in cancer for about two decades by an increasing number of molecular oncologists who contributed to deciphering the epigenetic codes and machinery and opened the road for a new generation of drugs now in clinical trials. However, the relevance of epigenetics to common diseases such as metabolic syndrome and cardiovascular disease was less conspicuous. This review focuses on converging data supporting the hypothesis that, in addition to « thrifty genotype » inheritance, individuals with metabolic syndrome (MetS) - combining disturbances in glucose and insulin metabolism, excess of predominantly abdominally distributed weight, mild dyslipidemia and hypertension, with the subsequent development of obesity, type 2 diabetes mellitus (T2D) and cardiovascular disease (CVD) - have suffered improper « epigenetic programming » during their fetal/postnatal development due to maternal inadequate nutrition and metabolic disturbances and also during their lifetime. Moreover, as seen for obesity and T2D, MetS tends to appear earlier in childhood, to be more severe from generation to generation and to affect more pregnant women. Thus, in addition to maternal effects, MetS patients may display « transgenerational effects » via the incomplete erasure of epigenetic marks endured by their parents and grandparents. We highlight the susceptibility of epigenetic mechanisms controlling gene expression to environmental influences due to their inherent malleability, emphasizing the participation of transposable elements and the potential role of imprinted genes during critical time windows in epigenetic programming, from the very beginning of development throughout life. Increasing our understanding on epigenetic patterns significance and small molecules (nutrients, drugs) that reverse epigenetic (in)activation should provide us with the means to « unlock » silenced (enhanced) genes, and to « convert » the obsolete human thrifty genotype into a « squandering » phenotype.
Article body
L’épidémie de syndrome métabolique
Le syndrome métabolique comprend un ensemble de troubles métaboliques incluant une résistance à l’insuline, une diminution du HDL-cholestérol, une hypertriglycéridémie, un surpoids abdominal, associés à une hypertension, un état inflammatoire et un état thrombotique. Les complications du syndrome métabolique (obésité, diabète de type 2 [DM2], maladies cardiovasculaires et cancers) ont un impact de plus en plus préoccupant sur la santé publique [1]. Depuis plus d’une décennie, la notion que ce type de maladies prend ses racines au cours de périodes cruciales de la programmation foetale et postnatale est largement étayée [2].
Une proportion grandissante de femmes (14 % à 27 %) débutent leur grossesse en état de surcharge pondérale. Si les effets du diabète gestationnel ont été bien étudiés, les effets sur la « programmation foetale » d’un syndrome métabolique chez la mère, associé à une alimentation déséquilibrée et à des troubles métaboliques, sont encore largement méconnus [3]. Par ailleurs, en cas de petit poids de naissance, une croissance postnatale précoce rapide (catch-up growth) peut être délétère, et il semble bien que l’effet le plus délétère (poids et longévité) soit consécutif à une trajectoire de croissance chaotique [4].
Une revue récente établit un bilan critique des travaux réalisés, essentiellement chez l’animal, sur l’étude de la « programmation foetale » [5]. Si la majorité des travaux ont porté sur l’étude des conséquences d’une restriction protéique pendant la gestation, ce sont pourtant les études s’intéressant aux effets d’un régime hypergras et/ou riche en hydrates de carbone qui, bien que moins nombreuses, semblent correspondre le mieux aux caractéristiques de l’épidémie actuelle de syndrome métabolique.
Régulation de l’expression génique par les modifications épigénétiques
Tous nos tissus contiennent les mêmes 30 000 gènes et pourtant, dans un tissu donné et à un stade donné, tous ne s’expriment pas : un « code épigénétique » permet à certains gènes d’être actifs (plus ou moins), alors que d’autres restent silencieux, de manière transitoire ou permanente ((→) m/s 2005, n° 4, p. 384 et 390).
Le code épigénétique comprend plusieurs strates interconnectées et interdépendantes : le code de la méthylation de l’ADN, le code des histones (acétylation, méthylation, phosphorylation…) et celui des co-activateurs et co-répresseurs. Ces codes sont mis en place et interprétés par une machinerie complexe d’enzymes et de co-facteurs qui assurent un remodelage adéquat de la chromatine autour des gènes et son accessibilité aux facteurs de transcription. Chez l’adulte, la modification des profils de méthylation de l’ADN au cours des processus de différenciation n’ont été décrits à ce jour que pour quelques gènes, mais il est clair que ce type de processus s’applique nécessairement à tous les gènes contrôlés au cours du développement et de la différenciation. Ainsi, on observe une stricte corrélation entre la déméthylation du promoteur de la leptine, son expression et la différenciation du pré-adipocyte en adipocyte [6, 7].
Contrairement aux mutations dans la séquence de l’ADN qui sont irréversibles, les modifications épigénétiques sont, en principe, instables et réversibles. Elles ont ainsi un caractère transitoire dans la vie courante : au cours de la journée, l’expression des gènes est modulée par les signaux moléculaires des rythmes circadiens, ainsi que par les signaux nutritionnels, au gré de la diversité de l’ensemble des stimulus environnementaux.
Enfin, des altérations de la méthylation de l’ADN s’accumulent avec l’âge, qui peuvent rendre compte de l’accentuation des symptômes du syndrome métabolique chez les individus âgés [3]. Des études menées chez la souris ApoE-/-, génétiquement prédisposée à l’athérosclérose, montrent que des profils épigénétiques anormaux sont détectables dans l’aorte bien avant l’apparition de lésions vasculaires détectables [8]. De plus, les altérations de la méthylation de l’ADN sont accentuées si les souris ApoE-/- sont alimentées avec un régime de type occidental, ou « caféteria » [9].
Programmation épigénétique au cours du développement foetoplacentaire et postnatal
En réponse à des programmes génétiques et épigénétiques orchestrés avec précision, les tissus et les organes sont façonnés, alternant prolifération, différenciation et apoptose. Plusieurs exemples de régulation épigénétique en relation avec le statut nutritionnel au cours de la vie utérine ou de la période postnatale ont ainsi été mis en évidence.
Les défauts de croissance foetoplacentaire sont associés à des altérations épigénétiques chez le rat. Au niveau du foie, par exemple, on observe une altération de la méthylation de l’ADN (hypométhylation) et de l’acétylation des histones en relation avec des modifications du métabolisme hépatique monocarboné de la voie des folates-méthionine [10] (Figure 1). L’inadéquation entre les apports (qualitatifs et quantitatifs) de nutriments ou de métabolites et les besoins précis de ces processus, au décours de l’espace/temps limité qui leur est dévolu, peut aboutir à des altérations des processus homéostasiques liées à des modifications épigénétiques instables et potentiellement réversibles, mais aussi à une situation de « non-retour » due à des altérations irréversibles des processus de différenciation et d’organogenèse, avec un développement structurel et fonctionnel anormal, voire une absence de certains types cellulaires spécialisés.
Figure 1
Métabolisme monocarboné de la voie des folates et de la méthionine.
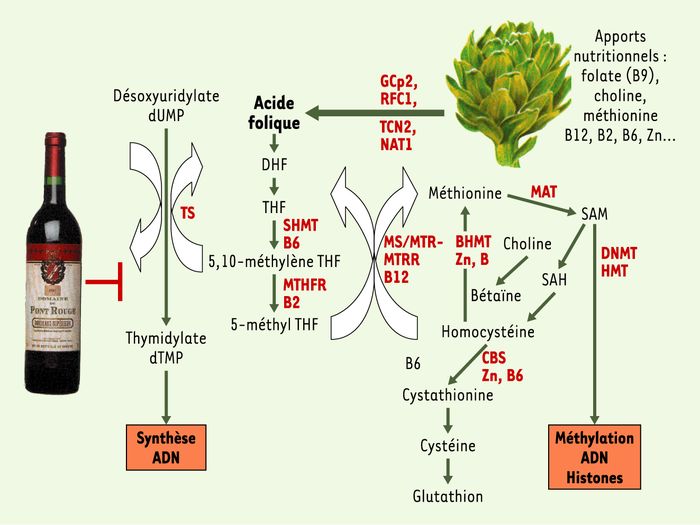
La voie de métabolisation des folates et de la méthionine, ou métabolisme monocarboné, a pour fonction le transfert d’unités à un carbone pour la synthèse de l’ADN et pour la méthylation de l’ADN et de protéines, tout en dépendant des apports nutritionnels. La choline, la méthionine, les vitamines B12, B2, B6, et B9 (ou acide folique), le zinc et d’autres micronutriments sont absorbés, modifiés et transportés par les produits de plusieurs gènes (GCP2, TCN2, RFC1, NAT1…). Le métabolisme des folates produit une source de groupements méthyl pour la conversion de l’homocystéine en méthionine, puis, via la MAT, aboutit à la production de S-adénosylméthionine (SAM), un donneur universel de groupements méthyl pour de nombreuses réactions biologiques et la synthèse de novo de désoxynucléotide triphosphate. La méthionine est régénérée à partir de l’homocystéine par la méthionine synthétase. La réaction de la méthionine synthétase est centrale pour l’approvisionnement en précurseurs métaboliques pour la méthylation de l’ADN (méthionine et SAM) et la synthèse de l’ADN (THF et 5,10-méthylène THF). La MTHFR catalyse la réduction de 5,10-méthylène THF en 5-méthyl THF. En raison de l’interdépendance métabolique entre la choline, la méthionine, les vitamines B12, B6, B2, l’acide folique et le zinc, tout excès ou déficience endogène ou exogène de ces molécules peut affecter l’approvisionnement en groupements méthyl et perturber les priorités métaboliques des autres molécules. La consommation excessive d’alcool diminue les taux de SAM, entraîne une hyperméthylation de certains gènes suppresseurs de tumeurs et une hypométhylation du génome. La déplétion en folates alimentaires diminue la méthylation de l’ADN.
Par ailleurs, la leptine, qui contrôle l’équilibre énergétique par la modulation électrophysiologique des neurones responsables de la régulation positive (NPY/AgRP, neuropeptide Y/Agouti related protein) ou négative (POMC/CART, pro-opiomélanocortine/cocaine- and amphetamine-regulated transcript ; αMSH, melanocyte-stimulating hormoneα) de la prise alimentaire, a également un rôle trophique sur certains neurones de l’hypothalamus impliqués dans la réponse aux aliments, et contribue à leur plasticité. Cependant, ce rôle primordial ne s’exerce que pendant une fenêtre très étroite de la période néonatale ; au-delà de ces limites, la maturation non achevée est irrécupérable [11].
Enfin, le taux d’apoptose pancréatique physiologique en période postnatale est augmenté chez les rats dont la mère a subi une restriction protéique durant la gestation, ce qui entraîne une diminution de la masse de cellules β du pancréas et perturbe, à la génération suivante, l’adaptation du pancréas endocrine [12]. De même, bien qu’il ne s’agisse plus d’une restriction, l’altération des fonctions pancréatiques (comme l’hyperinsulinisme) consécutive à un régime riche en hydrates de carbones se répercute sur le développement du pancréas à la deuxième génération, en dehors de tout stimulus nutritionnel délétère [13]. Il est néanmoins particulièrement intrigant de constater que des causes diamétralement opposées, sous-nutrition par une restriction protéique foetale et/ou postnatale ou surnutrition par un régime riche en hydrates de carbones ou en graisses, qui se traduisent par une diminution des flux sanguins foetoplacentaires et compromettent la croissance de l’embryon, aient les mêmes conséquences à long terme.
Un état épigénomique particulier peut également être mis en place par une programmation comportementale pendant une étroite fenêtre, et être potentiellement réversible. Ainsi, une augmentation des soins maternels à des rats nouveau-nés se traduit par une meilleure réponse au stress à l’âge adulte. L’équipe de Michael Meaney a récemment démontré que la méthylation du promoteur du gène du récepteur des glucocorticoïdes (RG) dans l’hippocampe est différente entre les petits de mères prodigant un bon niveau de soins maternels (mères HLG, high licking-grooming) et les petits de mères LLG (low licking-grooming) [14]. Ces différences apparaissent dès la première semaine de vie, avec une déméthylation caractéristique pour les petits de mères HLG. Elles persistent jusqu’à l’âge adulte et sont associées à une modification de l’acétylation des histones et à une augmentation du facteur de transcription NGFI-A (nerve growth factor-inducible protein A), qui se fixe sur le RG, avec pour résultat une augmentation de l’expression du récepteur. Ces modifications épigénétiques ne sont pas toujours totalement verrouillées. Ainsi, une injection au niveau central de trichostatine A, un inhibiteur d’histone désacétylase (HDAC), efface les différences épigénétiques entre les groupes d’animaux et supprime ainsi, chez l’adulte, les conséquences d’un comportement maternel postnatal inadéquat [14].
Gènes soumis à empreinte et croissance foetoplacentaire et postnatale
Les gènes soumis à empreinte parentale sont caractérisés par une expression mono-allélique : seul l’allèle paternel ou maternel s’exprime. L’autre allèle, intact, est « éteint » par des modifications épigénétiques. L’apposition du sceau parental a lieu au cours de la gamétogenèse. La complexité de la régulation au niveau des domaines soumis à empreinte, comportant à la fois des gènes à expression maternelle et des gènes à expression paternelle, peut les rendre particulièrement sensibles aux facteurs environnementaux et aux nutriments.
L’empreinte parentale ((→) m/s 2005, n° 4, p. 390) est rarement un phénomène de tout ou rien. L’expression mono-allélique des gènes soumis à empreinte parentale varie en effet d’un individu à l’autre et, pour un même individu, d’un tissu à l’autre, ainsi qu’au cours de l’embryogenèse, du développement et du vieillissement, voire dans certaines situations pathologiques comme le cancer ou l’athérosclérose. Ainsi, les modifications épigénétiques associées à l’empreinte pourraient se comporter comme un système tampon, une sorte de rhéostat, permettant une adaptation rapide aux conditions environnementales, grâce aux gènes s’exprimant de façon mono-allélique, en leur imposant le silence (allèle actif) ou, à l’inverse, en réactivant leur expression (allèle inactif) [15-18].
Nous n’avons pas encore la preuve formelle de l’implication de gènes soumis à empreinte parentale dans les processus d’adaptation de l’espèce à son environnement, mais un faisceau d’arguments vient étayer cette hypothèse (Encadré).
Gènes soumis à empreinte et croissance foetoplacentaire : apport et demande en nutriments
En accord avec la théorie du conflit entre les sexes, plusieurs gènes à expression maternelle contrôlent négativement la croissance foetale et placentaire, alors que plusieurs gènes à expression paternelle les contrôlent positivement (Figure 3). Grâce à des gènes soumis à empreinte appartenant aux familles de gènes active aminoacid transport system A, solute carrier ou organic cation transporter, l’empreinte parentale intervient dans la capacité de transport des nutriments, dans la régulation des interactions entre différents types cellulaires au niveau des interfaces foetomaternelles et contribue à la croissance du placenta et à sa résistance vasculaire, en contrôlant l’apport en nutriments. Dans les organes foetaux, les gènes soumis à empreinte contrôlent la demande en nutriments en réglant le taux de croissance des tissus foetaux [25] (Figure 3A).
Dans le placenta, la régulation de l’expression des gènes implique majoritairement une méthylation des histones qui semble indépendante de la méthylation de l’ADN [26]. Celle-ci est d’ailleurs réduite dans le placenta par rapport aux tissus somatiques (Figure 2), ce qui pourrait rendre compte d’une plus grande labilité épigénétique aux facteurs environnementaux des gènes placentaires soumis à empreinte.
Figure 2
Impact des nutriments ou des interventions nutritionnelles sur la reprogrammation de la méthylation de l’ADN pendant le développement et au cours de la vie.
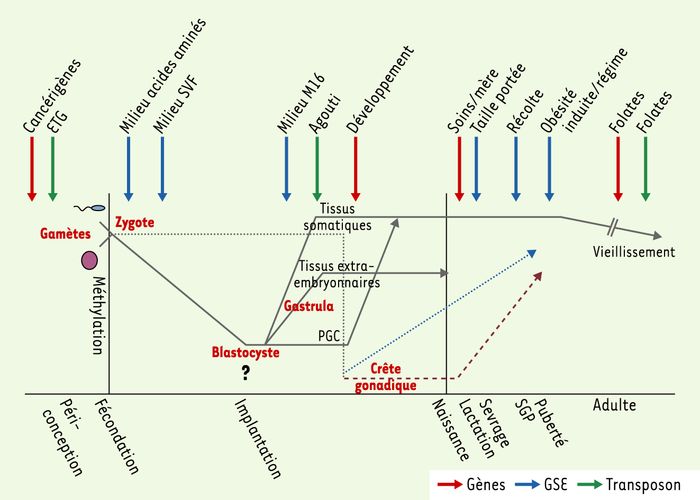
Après la fécondation, les génomes paternel et maternel du zygote subissent une rapide déméthylation au niveau des séquences codantes (gènes) et des séquences répétées (éléments transposables) (ligne pleine), à l’exception des marques épigénétiques des gènes soumis à empreinte et de certaines séquences répétées comme les transposons murins de type IAP (intracisternal A particle), qui semblent aussi réfractaires, dans une certaine mesure, à cette déméthylation. Une méthylation active de novo se met en route après l’implantation, à des degrés variables selon la partie de l’embryon concernée. Après l’implantation, l’ensemble du génome devient hyperméthylé grâce à une intense activité de méthylation de novo, alors que le génome des cellules extra-embryonnaires reste hypométhylé [26]. La méthylation « parent-spécifique » de l’empreinte parentale échappe au processus de déméthylation et de méthylation de novo (ligne brisée). Les empreintes des gènes soumis à empreinte parentale sont effacées avant que les cellules germinales primordiales (PGC) n’atteignent la crête gonadique, remises en place de manière appropriée pendant la gamétogenèse mâle et femelle et achevées pendant la période de croissance lente (SGP), avant la puberté (ligne brisée bleue pour le mâle, ligne pointillée rouge pour la femelle). Chez le mâle, l’acquisition des profils de méthylation commence avant la naissance dans les prospermatogonies et ne s’achève pour de nombreuses séquences qu’après la naissance, avant la fin du stade pachytène de la méiose. Contrairement au mâle, dans les cellules germinales femelles, la plus grande part de la méthylation gamétique se produit après la naissance, pendant la phase de croissance de l’ovocyte après le stade pachytène de la méiose. La SGP est associée à l’apparition du premier pool viable de spermatocytes et à l’initiation de la programmation de la méthylation des empreintes [29, 32]. À chaque stade de cette cascade de fluctuations épigénétiques - aussi bien pendant le développement foetal qu’au cours de la vie et du vieillissement - l’équilibre nutritionnel doit être « optimal ». L’impact, au niveau de différents types de séquences (gènes et séquences répétées), de régimes alimentaires déséquilibrés ou de certains nutriments chez l’animal - rat, souris, ovins essentiellement - sur les processus de programmation épigénétique au cours de la vie, pendant différentes périodes, avec leur transmission éventuelle d’une génération à l’autre, est aujourd’hui bien mis en évidence [23]. L’impact de différents types de nutriments ou de régimes est indiqué par des flèches, dans la fenêtre au cours de laquelle il a été observé. Ces différents impacts concernent soit des transposons (vert), soit des gènes (rouge), soit des gènes soumis à empreinte parentale (GSE) (bleu). Chaque flèche correspond à une référence citée dans cet article ou dans les références suivantes [14, 23, 33].
Figure 3
Effets des gènes soumis à empreinte sur l’acquisition des ressources par le foetus et le nourrisson.

A. Antagonisme des gènes soumis à empreinte parentale maternelle ou paternelle sur la régulation de la croissance du foetus (les gènes exprimés à partir de l’allèle maternel sont en rouge, ceux exprimés à partir de l’allèle paternel en bleu). La croissance foetale est promue par Igf2, par exemple, exprimé à partir de l’allèle paternel, qui peut signaler une augmentation de la demande au placenta, et accroître la capacité de transport des nutriments du placenta. Plusieurs autres gènes codant pour des transporteurs de nutriments (gènes Slc) sont également soumis à empreinte. Les gènes exprimés à partir de l’allèle maternel (comme Igf2r et Cdkn1c) peuvent, quant à eux, réduire l’approvisionnement ou la demande en nutriments. En accord avec la théorie du conflit d’intérêt entre les parents, les gènes à expression paternelle, comme Igf2, augmentent les apports de ressources vers le foetus, tandis que les gènes à expression maternelle, comme Cdkn1c, les freinent. L’invalidation de gènes à expression paternelle (Igf2P0, Peg1, Peg3) est ainsi associée à un retard de croissance intra-utérin, tandis que l’invalidation de gènes à expression maternelle (Ipl, H19, Grb10, Igf2r, Phlda2) est associée à une placentomégalie. B. Régulation de la croissance du nourrisson par les gènes soumis à empreinte parentale. Des gènes soumis à empreinte parentale peuvent également contrôler l’approvisionnement en ressources après la naissance, d’une part en agissant sur le cerveau de la mère pour réguler la production de lait, et, d’autre part, en agissant sur le cerveau de l’enfant, pour lui permettre, par exemple, de s’attacher au bout de sein. La tétée, et les comportements alimentaires en général, sont ainsi contrôlés par différents gènes soumis à empreinte, de même que certaines interactions émotionnelles avec la mère. Chez le nourrisson, ces gènes peuvent contribuer à la distribution des ressources pour permettre la croissance, la constitution des réserves de graisses et la maintenance de mécanismes homéostatiques tels que la régulation de la glycémie ou de la température. Bien que les effets de la plupart de ces gènes n’aient, pour l’heure, été mis en évidence que chez l’animal, il est probable qu’ils agissent de la même façon chez l’homme (d’après [19]).
Gènes soumis à empreinte et croissance postnatale
Les gènes soumis à empreinte parentale jouent également un rôle crucial pendant la période postnatale. Les deux exemples rapportés plus loin, concernant le complexe GNAS et les gènes Peg1 et Peg3, décrivent des mécanismes de solidarité entre les générations pouvant être mis en jeu face à l’abondance ou à la pénurie alimentaire [16] (Figure 3B).
Le complexe GNAS est un domaine soumis à empreinte codant, d’une part, pour le peptide stimulant la protéine Gsα (sous-unité régulatrice des protéines G stimulatrices), qui interagit avec l’adénylate cyclase, et, d’autre part, pour de nombreux autres transcrits s’exprimant de manière spécifique de tissu, soit à partir de l’allèle maternel (gonades, hypophyse, thyroïde, tubule proximal rénal et tissu adipeux), soit à partir de l’allèle paternel (tissu adipeux brun). Les phénotypes murins et humains associés à des altérations de ce locus montrent des effets opposés pour les pertes de fonction paternelle (diminution de l’adiposité, hypoglycémie, hypermétabolique, diminution de l’activité locomotrice, résistance à l’hormone parathyroïde) et maternelle (augmentation de l’adiposité). Les souriceaux ayant hérité de la délétion paternelle de l’isoforme extralarge de Gsα (Xlαs) ont des difficultés à têter, une hypoglycémie néonatale et une réduction du tissu adipeux brun. En revanche, la transmission maternelle est bénigne, le transcrit de cette isoforme n’étant pas produit à partir du chromosome maternel [21]. La régulation de l’homéostasie énergétique est également perturbée chez ces souriceaux. Les concentrations sanguines de glucose, d’insuline et de glucagon sont fortement diminuées, suggérant un dysfonctionnement pancréatique. La masse graisseuse interscapulaire, essentiellement composée de tissu adipeux brun, est considérablement réduite et montre une absence de particules lipidiques [21]. Enfin, les régions cérébrales exprimant l’isoforme Xlαs correspondent aux régions de contrôle de la vigilance, de l’insomnie, du sommeil et de l’homéostasie énergétique (hypothalamus), ainsi qu’à celles impliquées dans l’innervation des muscles des mâchoires. Ces régions d’expression sont à rapprocher du phénotype des souris invalidées : hypoactivité, difficultés à têter et troubles métaboliques. Ainsi, les souris n’exprimant plus Xlαs sont hypermétaboliques et maigres, et ont une sensibilité accrue à l’insuline. Ces résultats montrent que le complexe Gnas joue un rôle crucial dans l’adaptation métabolique postnatale du nouveau-né.
Les altérations de deux autres gènes soumis à empreinte, Peg1 et Peg3, qui s’expriment à partir de l’allèle paternel, montrent comment de tels gènes peuvent contrôler chez les différents partenaires, la mère et le foetus/nourrisson, de multiples aspects du développement foetal et postnatal. L’invalidation de l’allèle paternel de Peg3 entraîne, lorsqu’elle est portée par le foetus, une diminution de la taille du placenta, de la croissance foetale, de la capacité à têter et de la croissance postnatale et, enfin, de la température corporelle, ainsi qu’un retard du sevrage et de la puberté [22]. Cette mutation de l’allèle paternel de Peg3, lorsqu’elle est portée par la mère, compromet ses chances de reproduction, avec une diminution des soins maternels, de la prise de nourriture pendant la grossesse et de la production de lait. Les conséquences en sont, à nouveau pour le nourrisson, une diminution de la croissance postnatale et un retard du sevrage et de la puberté [22]. La synchronisation de ces traits co-adaptatifs, chez la mère et les petits, permet d’assurer que le petit qui aura tiré le maximum de sa mère sera lui-même bien approvisionné en nutriments, rendant les souriceaux femelles ainsi capables d’être à leur tour de « bonnes » mères apportant à leurs petits les meilleurs soins [22].
Par ailleurs, chez l’adulte, l’expression de Peg3 dans le tissu adipeux, ainsi que celle d’un autre gène soumis à empreinte parentale, Peg1, est fortement augmentée dans l’obésité induite par le régime alimentaire [27], et corrélée à la taille des adipocytes [28] : ce gène à expression paternelle pourrait donc favoriser la mise en réserve d’énergie.
Cependant, la démonstration formelle de l’implication des gènes soumis à empreinte dans les processus d’adaptation doit reposer sur une analyse détaillée des profils épigénétiques dans les régions clés des complexes de gènes soumis à empreinte parentale. En effet, l’augmentation ou la diminution de l’expression d’un gène soumis à empreinte parentale peut être attribuée soit à la variation de l’expression de l’allèle parental qui s’exprime normalement sans intervention de l’autre allèle, soit à une perte d’empreinte avec altération de région(s) différentiellement méthylée(s) (DMR) et expression (ou extinction) des deux allèles. Ce type d’analyse devrait permettre de discriminer ces deux possibilités, afin de montrer si ces états d’empreinte parentale altérés sont transmissibles à la génération suivante.
Effets transgénérationnels et adaptation
Une modification nutritionnelle pendant la période de lactation (lait riche en hydrates de carbone) peut entraîner la transmission à la seconde génération de l’hyper-insulinisme, phénotype acquis à la première génération [13]. Chez l’homme, des données épidémiologiques suédoises vont dans ce sens : un grand-père « bien nourri » pendant sa période prépubertaire « transmet » à ses petits-enfants un risque multiplié par 4 de développer un diabète de type 2 [29] (Figure 2). Enfin, la programmation par le comportement maternel persiste bien à l’âge adulte, et montre un effet transgénérationnel, puisqu’à leur tour les femelles « bien maternées » deviennent elles-mêmes de bonnes mères [14].
Les transposons et les gènes soumis à empreinte parentale pourraient être les supports épigénétiques des altérations transmises aux générations suivantes par des « effets transgénérationnels » [23, 30] (Figure 2). Un mécanisme épigénétique rendant compte de tels effets a été décrit sur les locus Agouti (A) et Axin. Chez le mutant Agouti viable yellow Avy, les changements de coloration du pelage et d’autres aspects phénotypiques (hyper-insulinisme, obésité, tumeurs) dépendent de la méthylation d’une séquence transposable IAP (intracisternal A particle) insérée à proximité du gène A. À la génération suivante, les phénotypes variables observés dans la descendance sont dus à l’effaçage incomplet de la modification épigénétique lorsque l’allèle Avy est transmis à travers la lignée germinale maternelle. Le même phénomène se produit pour la séquence IAP insérée dans le locus Axin, et montre que la transmission paternelle ou maternelle dépend du fond génétique [23]. Enfin, un régime riche en donneurs de méthyl pendant la gestation se traduit par une augmentation de la proportion des petits porteurs de séquences IAP méthylées.
Bien qu’aucun mécanisme analogue n’ait encore été décrit chez l’homme, mais en considérant qu’une importante proportion du génome humain est représentée par des transposons (> 40 %), on ne peut négliger l’impact substantiel, en termes de santé publique, des influences nutritionnelles précoces sur ces séquences.
Conclusions
L’objectif des recherches en cours est d’identifier les gènes concernés par les altérations épigénétiques liées à la nutrition et de déchiffrer le message des différents profils épigénétiques. La labilité des modifications épigénétiques modulables par l’alimentation, observées au niveau de certains gènes, mais aussi des transposons et des gènes soumis à empreinte parentale, de même que le rôle de ces gènes soumis à empreinte parentale dans la croissance foetale, placentaire et postnatale, ainsi que dans le développement cérébral, suggèrent que les gènes soumis à empreinte parentale sont les supports les plus plausibles de ces altérations épigénétiques liées à l’adaptation de l’individu - de l’espèce - à son alimentation. Les résultats obtenus chez l’animal et les essais cliniques en cours chez l’homme dans le cadre de traitement de tumeurs (avec par exemple des chimiothérapies à base de cisplatine, budésonide ou décitabine) laissent envisager, lorsque les mécanismes épigénétiques en jeu et les fenêtres critiques auront été décryptés, de nouvelles perspectives de prévention ou de traitement (Figure 4). Compte tenu des interrelations entre les différentes voies épigénétiques, la combinaison d’un médicament avec un régime ou des nutriments appropriés pourrait être plus efficace que le médicament isolé [31].
Figure 4
Facteurs métaboliques et environnementaux pouvant influencer les profils d’acétylation des histones et de méthylation de l’ADN.
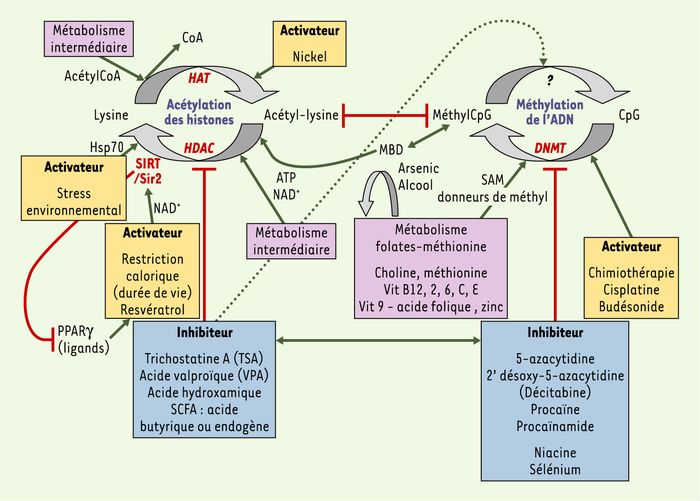
La méthylation de l’ADN et l’acétylation des histones sont les processus épigénétiques les mieux étudiés et pour lesquels des molécules capables d’interagir avec eux sont les mieux connues à ce jour : les facteurs métaboliques (en mauve) comprennent les concentrations intracellulaires d’acétylCoA, pour l’activité histone-acétylase (HAT), de NAD+, pour la désacétylase Sir2/SIRT1, d’ATP, pour la désacétylation des substrats chromatiniens par certaines histone-désacétylases (HDAC), et de donneurs de groupements méthyl (SAM, par exemple) apportés par la voie des folates-méthionine, pour l’activité des méthyltransférases de l’ADN (DNMT). De son côté, la réponse au stress (protéine de choc thermique HSP70) opère au sein de complexes HDAC, dans lesquels les HSP peuvent jouer un rôle dans la désacétylation des substrats chromatiniens. L’acide butyrique change quant à lui la structure de la chromatine (acétylation et phosphorylation des histones). Enfin, les micronutriments dont le défaut ou l’excès peuvent perturber les processus épigénétiques incluent le zinc, le sélénium, l’arsenic, le nickel, le fer, la vitamine C et la niacine, un précurseur du NAD+. L’arsenic et l’alcool, quant à eux, déplètent les pools de donneurs de groupements méthyle. Des lignées cellulaires de patients ou des modèles animaux ont permis de tester la réversibilité des modifications épigénétiques en utilisant différents inhibiteurs (en bleu) ou activateurs (en jaune) ((→) m/s 2005, n° 4, p. 405) : inducteurs de la méthylation (cisplatine et budésonide) ; inhibiteurs des méthyltransférases de l’ADN (5-azacytidine, 2’ désoxy-5-azacytidine, procaïnamide - un anti-arhytmique, procaïne - un anesthésique) ; inhibiteurs des histone-désacétylases (trichostatine A [TSA], acide valproïque [VPA] - un anti-épileptique) ; activateurs de la désacétylase SIRT1/Sir2 (resvératrol - un polyphénol du raisin - et restriction calorique). La sirtuine 1 est le médiateur des effets sur l’allongement de la durée de vie lié à la restriction calorique, par la mobilisation des graisses dans l’adipocyte et l’inhibition de PPARγ.
Appendices
Remerciements
Ce travail a été soutenu financièrement par une bourse Nestlé (AV), une bourse des laboratoires Fournier-Pharma (CGK) et des subventions de l’Inra, l’Inserm (ATC-Nutrition, PRNH), l’Association française des Diabétiques et l’Institut Benjamin-Delessert.
Références
- 1. Grundy SM. Obesity, metabolic syndrome, and cardiovascular disease. J Clin Endocrinol Metab 2004 ; 89 : 2595-600.
- 2. Ozanne SE, Fernandez-Twinn D, Hales CN. Fetal growth and adult diseases. Semin Perinatol 2004 ; 28 : 81-7.
- 3. Issa JP. Epigenetic variation and human disease. J Nutr 2002 ; 132 : 2388S-2392S.
- 4. Ozanne SE, Hales CN. Lifespan: Catch-up growth and obesity in male mice. Nature 2004 ; 427 : 411-2.
- 5. Armitage JA, Khan IY, Taylor PD, et al. Developmental programming of the metabolic syndrome by maternal nutritional imbalance : how strong is the evidence from experimental models in mammals ? J Physiol 2004 ; 561 : 355-77.
- 6. Melzner I, Scott V, Dorsch K, et al. Leptin gene expression in human preadipocytes is switched on by maturation-induced demethylation of distinct CpGs in its proximal promoter. J Biol Chem 2002 ; 277 : 45420-7.
- 7. Yokomori N, Tawata M, Onaya T. DNA demethylation modulates mouse leptin promoter activity during the differentiation of 3T3-L1 cells. Diabetologia 2002 ; 45 : 140-8.
- 8. Lund G, Andersson L, Lauria M, et al. DNA methylation polymorphisms precede any histological sign of atherosclerosis in mice lacking apolipoprotein E. J Biol Chem 2004 ; 279 : 29147-54.
- 9. Hiltunen MO, Turunen MP, Hakkinen TP, et al. DNA hypomethylation and methyltransferase expression in atherosclerotic lesions. Vasc Med 2002 ; 7 : 5-11.
- 10. MacLennan NK, James SJ, Melnyk S, et al. Uteroplacental insufficiency alters DNA methylation, one-carbon metabolism, and histone acetylation in IUGR rats. Physiol Genomics 2004 ; 18 : 43-50.
- 11. Elmquist JK, Flier JS. Neuroscience. The fat-brain axis enters a new dimension. Science 2004 ; 304 : 63-4.
- 12. Blondeau B, Avril I, Duchene B, Breant B. Endocrine pancreas development is altered in foetuses from rats previously showing intra-uterine growth retardation in response to malnutrition. Diabetologia 2002 ; 45 : 394-401.
- 13. Srinivasan M, Aalinkeel R, Song F, Patel MS. Programming of islet functions in the progeny of hyperinsulinemic/obese rats. Diabetes 2003 ; 52 : 984-90.
- 14. Weaver IC, Cervoni N, Champagne FA, et al. Epigenetic programming by maternal behavior. Nat Neurosci 2004 ; 7 : 847-54.
- 15. Pembrey M. Imprinting and transgenerational modulation of gene expression: Human growth as a model. Acta Genet Med Gemellol (Roma) 1996 ; 45 : 111-25.
- 16. Junien C. L’empreinte parentale : de la guerre des sexes à la solidarité entre générations. Med Sci (Paris) 2000 ; 3 : 336-344.
- 17. Beaudet AL, Jiang YH. A rheostat model for a rapid and reversible form of imprinting-dependent evolution. Am J Hum Genet 2002 ; 70 : 1389-97.
- 18. Young LE. Imprinting of genes and the Barker hypothesis. Twin Res 2001 ; 4 : 307-17.
- 19. Constancia M, Kelsey G, Reik W. Resourceful imprinting. Nature 2004 ; 432 : 53-7.
- 20. Keverne EB. Genomic imprinting and the maternal brain. Prog Brain Res 2001 ; 133 : 279-85.
- 21. Plagge A, Gordon E, Dean W, et al. The imprinted signaling protein XL alpha s is required for postnatal adaptation to feeding. Nat Genet 2004 ; 36 : 818-26.
- 22. Curley JP, Barton S, Surani A, Keverne EB. Coadaptation in mother and infant regulated by a paternally expressed imprinted gene. Proc R Soc Lond B Biol Sci 2004 ; 271 : 1303-9.
- 23. Waterland RA, Jirtle RL. Early nutrition, epigenetic changes at transposons and imprinted genes, and enhanced susceptibility to adult chronic diseases. Nutrition 2004 ; 20 : 63-8.
- 24. Delrue MA, Michaud JL. Fat chance: Genetic syndromes with obesity. Clin Genet 2004 ; 66 : 83-93.
- 25. Reik W, Constancia M, Fowden A, et al. Regulation of supply and demand for maternal nutrients in mammals by imprinted genes. J Physiol 2003 ; 547 : 35-44.
- 26. Umlauf D, Goto Y, Cao R, et al. Imprinting along the Kcnq1 domain on mouse chromosome 7 involves repressive histone methylation and recruitment of Polycomb group complexes. Nat Genet 2004 ; 36 : 1296-300.
- 27. Moraes RC, Blondet A, Birkenkamp-Demtroeder K, et al. Study of the alteration of gene expression in adipose tissue of diet-induced obese mice by microarray and reverse transcription-polymerase chain reaction analyses. Endocrinology 2003 ; 144 : 4773-82.
- 28. Takahashi M, Kamei Y, Ezaki O. Mest/Peg1 imprinted gene enlarges adipocytes and is a marker of adipocyte size. Am J Physiol Endocrinol Metab 2005 ; 288 : E117-24.
- 29. Kaati G, Bygren LO, Edvinsson S. Cardiovascular and diabetes mortality determined by nutrition during parents’ and grandparents’ slow growth period. Eur J Hum Genet 2002 ; 10 : 682-8.
- 30. Pembrey ME. Time to take epigenetic inheritance seriously. Eur J Hum Genet 2002 ; 10 : 669-71.
- 31. Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004 ; 429 : 457-63.
- 32. Kelly TL, Trasler JM. Reproductive epigenetics. Clin Genet 2004 ; 65 : 247-60.
- 33. Mann MR, Lee SS, Doherty AS, et al. Selective loss of imprinting in the placenta following preimplantation development in culture. Development 2004 ; 131 : 3727-35.
List of figures
Figure 1
Métabolisme monocarboné de la voie des folates et de la méthionine.
La voie de métabolisation des folates et de la méthionine, ou métabolisme monocarboné, a pour fonction le transfert d’unités à un carbone pour la synthèse de l’ADN et pour la méthylation de l’ADN et de protéines, tout en dépendant des apports nutritionnels. La choline, la méthionine, les vitamines B12, B2, B6, et B9 (ou acide folique), le zinc et d’autres micronutriments sont absorbés, modifiés et transportés par les produits de plusieurs gènes (GCP2, TCN2, RFC1, NAT1…). Le métabolisme des folates produit une source de groupements méthyl pour la conversion de l’homocystéine en méthionine, puis, via la MAT, aboutit à la production de S-adénosylméthionine (SAM), un donneur universel de groupements méthyl pour de nombreuses réactions biologiques et la synthèse de novo de désoxynucléotide triphosphate. La méthionine est régénérée à partir de l’homocystéine par la méthionine synthétase. La réaction de la méthionine synthétase est centrale pour l’approvisionnement en précurseurs métaboliques pour la méthylation de l’ADN (méthionine et SAM) et la synthèse de l’ADN (THF et 5,10-méthylène THF). La MTHFR catalyse la réduction de 5,10-méthylène THF en 5-méthyl THF. En raison de l’interdépendance métabolique entre la choline, la méthionine, les vitamines B12, B6, B2, l’acide folique et le zinc, tout excès ou déficience endogène ou exogène de ces molécules peut affecter l’approvisionnement en groupements méthyl et perturber les priorités métaboliques des autres molécules. La consommation excessive d’alcool diminue les taux de SAM, entraîne une hyperméthylation de certains gènes suppresseurs de tumeurs et une hypométhylation du génome. La déplétion en folates alimentaires diminue la méthylation de l’ADN.
Figure 2
Impact des nutriments ou des interventions nutritionnelles sur la reprogrammation de la méthylation de l’ADN pendant le développement et au cours de la vie.
Après la fécondation, les génomes paternel et maternel du zygote subissent une rapide déméthylation au niveau des séquences codantes (gènes) et des séquences répétées (éléments transposables) (ligne pleine), à l’exception des marques épigénétiques des gènes soumis à empreinte et de certaines séquences répétées comme les transposons murins de type IAP (intracisternal A particle), qui semblent aussi réfractaires, dans une certaine mesure, à cette déméthylation. Une méthylation active de novo se met en route après l’implantation, à des degrés variables selon la partie de l’embryon concernée. Après l’implantation, l’ensemble du génome devient hyperméthylé grâce à une intense activité de méthylation de novo, alors que le génome des cellules extra-embryonnaires reste hypométhylé [26]. La méthylation « parent-spécifique » de l’empreinte parentale échappe au processus de déméthylation et de méthylation de novo (ligne brisée). Les empreintes des gènes soumis à empreinte parentale sont effacées avant que les cellules germinales primordiales (PGC) n’atteignent la crête gonadique, remises en place de manière appropriée pendant la gamétogenèse mâle et femelle et achevées pendant la période de croissance lente (SGP), avant la puberté (ligne brisée bleue pour le mâle, ligne pointillée rouge pour la femelle). Chez le mâle, l’acquisition des profils de méthylation commence avant la naissance dans les prospermatogonies et ne s’achève pour de nombreuses séquences qu’après la naissance, avant la fin du stade pachytène de la méiose. Contrairement au mâle, dans les cellules germinales femelles, la plus grande part de la méthylation gamétique se produit après la naissance, pendant la phase de croissance de l’ovocyte après le stade pachytène de la méiose. La SGP est associée à l’apparition du premier pool viable de spermatocytes et à l’initiation de la programmation de la méthylation des empreintes [29, 32]. À chaque stade de cette cascade de fluctuations épigénétiques - aussi bien pendant le développement foetal qu’au cours de la vie et du vieillissement - l’équilibre nutritionnel doit être « optimal ». L’impact, au niveau de différents types de séquences (gènes et séquences répétées), de régimes alimentaires déséquilibrés ou de certains nutriments chez l’animal - rat, souris, ovins essentiellement - sur les processus de programmation épigénétique au cours de la vie, pendant différentes périodes, avec leur transmission éventuelle d’une génération à l’autre, est aujourd’hui bien mis en évidence [23]. L’impact de différents types de nutriments ou de régimes est indiqué par des flèches, dans la fenêtre au cours de laquelle il a été observé. Ces différents impacts concernent soit des transposons (vert), soit des gènes (rouge), soit des gènes soumis à empreinte parentale (GSE) (bleu). Chaque flèche correspond à une référence citée dans cet article ou dans les références suivantes [14, 23, 33].
Figure 3
Effets des gènes soumis à empreinte sur l’acquisition des ressources par le foetus et le nourrisson.
A. Antagonisme des gènes soumis à empreinte parentale maternelle ou paternelle sur la régulation de la croissance du foetus (les gènes exprimés à partir de l’allèle maternel sont en rouge, ceux exprimés à partir de l’allèle paternel en bleu). La croissance foetale est promue par Igf2, par exemple, exprimé à partir de l’allèle paternel, qui peut signaler une augmentation de la demande au placenta, et accroître la capacité de transport des nutriments du placenta. Plusieurs autres gènes codant pour des transporteurs de nutriments (gènes Slc) sont également soumis à empreinte. Les gènes exprimés à partir de l’allèle maternel (comme Igf2r et Cdkn1c) peuvent, quant à eux, réduire l’approvisionnement ou la demande en nutriments. En accord avec la théorie du conflit d’intérêt entre les parents, les gènes à expression paternelle, comme Igf2, augmentent les apports de ressources vers le foetus, tandis que les gènes à expression maternelle, comme Cdkn1c, les freinent. L’invalidation de gènes à expression paternelle (Igf2P0, Peg1, Peg3) est ainsi associée à un retard de croissance intra-utérin, tandis que l’invalidation de gènes à expression maternelle (Ipl, H19, Grb10, Igf2r, Phlda2) est associée à une placentomégalie. B. Régulation de la croissance du nourrisson par les gènes soumis à empreinte parentale. Des gènes soumis à empreinte parentale peuvent également contrôler l’approvisionnement en ressources après la naissance, d’une part en agissant sur le cerveau de la mère pour réguler la production de lait, et, d’autre part, en agissant sur le cerveau de l’enfant, pour lui permettre, par exemple, de s’attacher au bout de sein. La tétée, et les comportements alimentaires en général, sont ainsi contrôlés par différents gènes soumis à empreinte, de même que certaines interactions émotionnelles avec la mère. Chez le nourrisson, ces gènes peuvent contribuer à la distribution des ressources pour permettre la croissance, la constitution des réserves de graisses et la maintenance de mécanismes homéostatiques tels que la régulation de la glycémie ou de la température. Bien que les effets de la plupart de ces gènes n’aient, pour l’heure, été mis en évidence que chez l’animal, il est probable qu’ils agissent de la même façon chez l’homme (d’après [19]).
Figure 4
Facteurs métaboliques et environnementaux pouvant influencer les profils d’acétylation des histones et de méthylation de l’ADN.
La méthylation de l’ADN et l’acétylation des histones sont les processus épigénétiques les mieux étudiés et pour lesquels des molécules capables d’interagir avec eux sont les mieux connues à ce jour : les facteurs métaboliques (en mauve) comprennent les concentrations intracellulaires d’acétylCoA, pour l’activité histone-acétylase (HAT), de NAD+, pour la désacétylase Sir2/SIRT1, d’ATP, pour la désacétylation des substrats chromatiniens par certaines histone-désacétylases (HDAC), et de donneurs de groupements méthyl (SAM, par exemple) apportés par la voie des folates-méthionine, pour l’activité des méthyltransférases de l’ADN (DNMT). De son côté, la réponse au stress (protéine de choc thermique HSP70) opère au sein de complexes HDAC, dans lesquels les HSP peuvent jouer un rôle dans la désacétylation des substrats chromatiniens. L’acide butyrique change quant à lui la structure de la chromatine (acétylation et phosphorylation des histones). Enfin, les micronutriments dont le défaut ou l’excès peuvent perturber les processus épigénétiques incluent le zinc, le sélénium, l’arsenic, le nickel, le fer, la vitamine C et la niacine, un précurseur du NAD+. L’arsenic et l’alcool, quant à eux, déplètent les pools de donneurs de groupements méthyle. Des lignées cellulaires de patients ou des modèles animaux ont permis de tester la réversibilité des modifications épigénétiques en utilisant différents inhibiteurs (en bleu) ou activateurs (en jaune) ((→) m/s 2005, n° 4, p. 405) : inducteurs de la méthylation (cisplatine et budésonide) ; inhibiteurs des méthyltransférases de l’ADN (5-azacytidine, 2’ désoxy-5-azacytidine, procaïnamide - un anti-arhytmique, procaïne - un anesthésique) ; inhibiteurs des histone-désacétylases (trichostatine A [TSA], acide valproïque [VPA] - un anti-épileptique) ; activateurs de la désacétylase SIRT1/Sir2 (resvératrol - un polyphénol du raisin - et restriction calorique). La sirtuine 1 est le médiateur des effets sur l’allongement de la durée de vie lié à la restriction calorique, par la mobilisation des graisses dans l’adipocyte et l’inhibition de PPARγ.