Abstracts
Résumé
Les études conduites au cours des vingt dernières années ont constitué un apport majeur pour la compréhension de la transformation tumorale. Elles ont ouvert un domaine nouveau de la génétique : l’étude de la prédisposition au cancer. Aujourd’hui, les situations les plus simples de prédisposition ont été identifiées : il s’agit d’altérations monogéniques s’accompagnant d’un risque tumoral élevé et de phénotypes spécifiques. Une quarantaine de gènes ont été identifiés, qui font l’objet d’une recherche par le biais de tests génétiques : l’indication de ces tests et la prise en charge ultérieure des sujets à risque doivent être réfléchies de façon collégiale, et des recommandations de prise en charge établies. Toutefois, un vrai défi à relever concerne l’identification des facteurs de prédispositions associés à des risques plus faibles, ou ayant un impact sur la réponse des patients aux traitements.
Summary
Studies performed during these last twenty years have had a major impact on the understanding of carcinogenesis. They have opened a new field : cancer genetic predisposition. At the present time, most of the cancer predispositions linked to the alteration of one gene, associated with a high risk of cancer and with a specific phenotype have been identified. About 40 genes have been identified and have led to genetic testing. The indication of genetic testing, the management of at risk patients need the establishment of guidelines. The next challenge is the identification of cancer predisposing genes associated with low risk or modifying the effect of treatment.
Article body
Le cancer est une maladie génétique de la cellule : il s’agit là d’un dogme que les vingt dernières années de recherche n’ont cessé de vérifier [1]. En effet, la transformation tumorale d’une cellule est un processus clonal, procédant par des étapes successives constituées, en général, de mutations ; la cellule ayant franchi l’une de ces étapes a acquis un avantage sélectif par rapport à ses congénères. Ces étapes permettent à la cellule d’acquérir capacité de survie et augmentation de la prolifération, à travers la perte du contrôle de l’arrêt du cycle, l’inhibition de l’apoptose et de la différenciation ou, encore, l’acquisition de capacités angiogéniques et métastatiques. Ces étapes cellulaires d’évolution clonale sont des mutations (ou des modifications épigénétiques) de gènes dont le produit participe dans une, voire plusieurs voies cellulaires impliquées dans les processus cités plus haut. Le nombre de mutations, ou plutôt le quota de mutations nécessaires à la transformation cellulaire est faible, et variable d’une tumeur à une autre : il est probablement de moins de 5 dans les tumeurs embryonnaires de l’enfant, et d’une dizaine dans les carcinomes de l’adulte.
Ces mutations peuvent être activatrices, conduisant à une hyperactivité de la protéine correspondante (amplification génique), à son expression constitutive (mutation faux-sens) ou à la création d’une nouvelle protéine (fusion génique). La mutation d’un seul allèle suffit à conférer un avantage sélectif à la cellule ; le gène cible de cette mutation est un gène « oncogène ». À l’inverse, il peut s’agir de mutations inactivatrices (stop, insertion ou délétion de quelques nucléotides, anomalies d’épissage, faux-sens, réarrangements de grande taille du gène…). Ces mutations sont, dans la majorité des cas, associées à une inactivation du second allèle tumoral, le plus souvent par perte de la région chromosomique contenant le gène en question, un phénomène mis en évidence par la recherche de perte d’hétérozygotie au niveau tumoral. Les gènes cibles de ces mutations inactivatrices sont des gènes « suppresseurs de tumeur ». L’acquisition de mutations dans les gènes oncogènes ou suppresseurs de tumeur peut être secondaire aux erreurs de réplication de l’ADN survenant lors de chaque division cellulaire ; elle est naturellement favorisée par l’exposition de la cellule aux agents mutagènes. On comprend ainsi que des mutations dans les gènes de réparation de l’ADN, mais aussi dans les gènes impliqués dans la stabilité des chromosomes, aient également un rôle clé, bien qu’indirect, dans le processus tumoral, en augmentant le taux de mutations dans les gènes oncogènes ou suppresseurs de tumeurs, et donc la probabilité de voir émerger un processus tumoral. Une grande partie de la recherche a aujourd’hui pour objectif d’identifier, dans les différents cancers, les gènes mutés (et donc les voies cellulaires altérées), ainsi que les gènes de stabilité (ou réparation) du génome. En 2004, près de 300 gènes (soit environ 1 % du génome), impliqués directement ou indirectement dans l’oncogenèse, ont été recensés par le Sanger Center : la majorité d’entre eux sont des oncogènes [2] (projet COSMIC : http://www.sanger.ac.uk/CGP/).
La notion de prédisposition génétique au(x) cancer(s) est une notion relative : elle correspond à une augmentation du risque de cancers, ou d’un cancer donné, chez une personne, par rapport au risque moyen observé dans la population générale. Cette augmentation peut être mesurée par le risque relatif ; autrement dit, si l’on revient au processus biologique de la transformation cellulaire, il s’agit de situations où le quota de mutations nécessaire à la transformation cellulaire est atteint plus rapidement. Ainsi, lorsqu’une mutation d’un gène clé du processus tumoral est constitutionnelle, une étape est déjà franchie. L’archétype en est la prédisposition au rétinoblastome, où l’inactivation des deux allèles du gène RB1 est nécessaire (mais peut-être pas suffisante) à la transformation tumorale des neuroblastes de la rétine du jeune enfant. Dans plus de 60 % des cas, l’atteinte est unilatérale et survient après l’âge de 12 mois. Dans la plupart de ces cas, les deux mutations ont été acquises. En revanche, dans près de 40 % des cas, l’atteinte est bilatérale et survient avant l’âge de 12 mois et, dans près de 10 % des cas (25 % des cas bilatéraux), l’un des deux parents a été atteint dans l’enfance. Chez ces enfants, l’une des deux mutations du gène RB1 est constitutionnelle, l’autre est somatique. Le gène RB1, cloné en 1986, est le premier gène de prédisposition aux cancers qui ait été identifié. Son identification a ouvert un nouveau champ de la génétique.
Prédispositions aux cancers identifiées
En moins de 20 ans, une quarantaine de gènes de prédisposition aux cancers ont été identifiés (Tableau I). Ils correspondent aux situations de prédisposition les plus simples à mettre en évidence, la partie émergée d’un iceberg que représenterait l’ensemble des prédispositions aux cancers. En effet, il s’agit soit de prédispositions transmises selon le mode dominant et associées à un risque tumoral élevé, conduisant alors souvent à une concentration familiale de cancers, soit de syndromes dont les manifestations désignent la prédisposition. Les archétypes de ces maladies sont les hamartomatoses et les maladies cassantes de l’ADN. Les gènes de ces deux groupes de prédisposition ont, dans la majorité des cas, été identifiés par étude de liaison génétique puis clonage positionnel, parfois par des approches gène-candidat.
Tableau I
Gènes de prédisposition aux cancers identifiés.
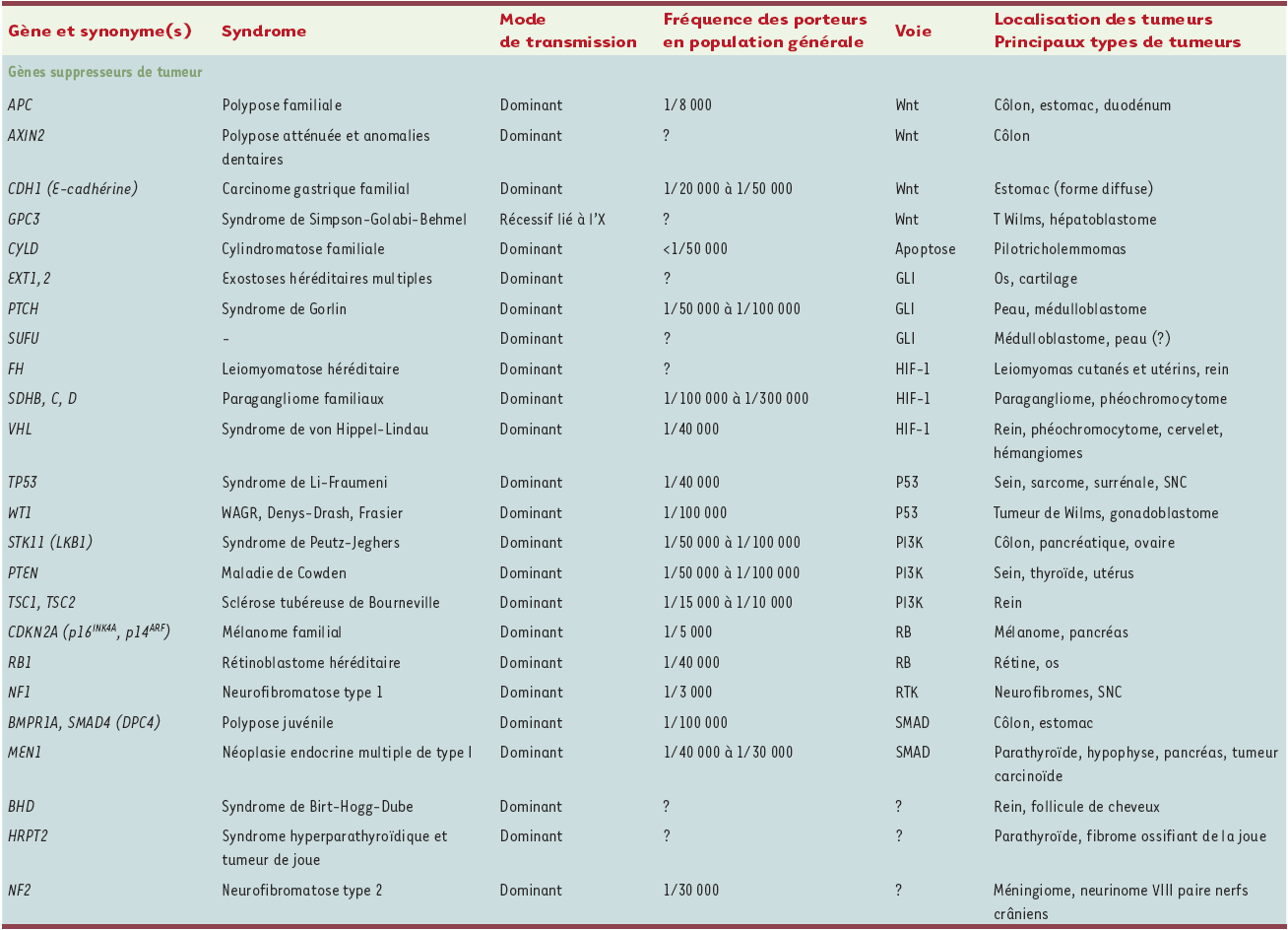
Tableau I (continuation)

La fréquence des porteurs dans la population générale est donnée à titre indicatif et est susceptible de varier d’un facteur 2, voire plus pour les fréquences très faibles. Lorsque la fréquence est inconnue (notée ?), elle est a priori très faible. WAGR : Wilms tumor-aniridia-genitourinary anomalies-mental retardation ; HNPCC : hereditary non polyposis carcinoma ; HIF-1 : hypoxemia induced factor-1 ; PI3K : phosphatidylinositol 3 kinase ; RB : protéine du rétinoblastome ; RTK : récepteurs des tyrosine kinases ; HNF1 : hepatocyte nuclear factor 1 ; BER : base excision repair ; CIN : instabilité chromosomique ; MMR : mismatch repair ; NER : nucleotide excision repair (complété d’après [1]).
L’un des grands succès des approches de liaison est l’identification des gènes de prédisposition aux cancers du sein et de l’ovaire, BRCA1 et BRCA2. Après des études de ségrégation ayant retenu un modèle dominant avec ses paramètres de fréquence génique et de pénétrance, la réunion d’une trentaine de familles sélectionnées a permis à Mary-Claire King, en 1990, de localiser BRCA1 en 17q21 [3]. Puis, dans le cadre d’un consortium de laboratoires, la position du gène recherché a pu être finement précisée, et a conduit à son identification [4, 5]. Quant à l’approche gène-candidat, un succès très élégant concerne l’identification des gènes MLH1, MSH2 et MSH6, dont les altérations sont associées à un risque élevé de cancer du côlon et de l’endomètre : alors que des régions du génome perdues de façon récurrente dans des adénocarcinomes du côlon de personnes s’inscrivant dans un syndrome HNPCC (hereditary non polyposis carcinoma) étaient recherchées par une approche de perte d’hétérozygotie de régions microsatellites, des pertes d’allèle, et non des gains d’allèle, ont été en réalité mises en évidence dans ce type de tumeurs, évoquant un défaut de réparation d’erreurs de réplication de l’ADN (phénotype MSI, microsatellites instable). Ces gains d’allèles étaient très semblables à ce qui était observé chez certaines bactéries ; les homologues humains des gènes altérés chez ces bactéries, en l’occurrence hMLH1, hMSH2 et hMSH6, ont été identifiés, et des mutations constitutionnelles de ces gènes recherchées et mises en évidence (pour revue, voir [6]).
Ces dernières années ont montré que les gènes de prédisposition aux cancers n’appartenaient pas à des voies cellulaires isolées, dont les altérations ne conduiraient qu’à des cancers. Ainsi, des anomalies du développement empruntent des voies communes à certaines prédispositions aux cancers : voies Sonic-Hedgehog-Gli, avec holoprosencéphalie et syndrome de Greig d’une part, et syndrome de Gorlin, d’autre part, syndrome associé à un risque majeur de carcinomes basocellulaires multiples et à un risque plus faible de médulloblastome (pour revue, voir [7]). De la même façon, les gènes de la cascade des TGF-β sont impliqués à la fois dans des anomalies vasculaires et des tumeurs digestives [8]. Une autre voie passionnante mérite d’être mentionnée, celle du facteur HIF-1 (hypoxemia induced factor-1) : il s’agit d’un gène clé de l’induction de l’angiogenèse, dont l’expression est finement régulée par différents gènes, dont le gène VHL à l’origine de la maladie de von Hippel Lindau. D’autres gènes, codant pour des enzymes du cycle de Krebs, ont été récemment impliqués dans certaines prédispositions aux cancers : gènes des sous-unités de la succinate déshydrogénase (SDH) et gène de la fumarate hydratase (FH), dont les altérations à l’état hétérozygote sont, respectivement, associées à un risque de paragangliomes et de cancer du rein. À l’état homozygote ou hétérozygote composite, ces altérations sont responsables d’anomalies graves du métabolisme et de troubles neurologiques (pour revue, voir [9]).
Il est difficile de faire une présentation synthétique et exhaustive de l’ensemble des prédispositions aux cancers identifiées aujourd’hui, en 2005 : faut-il retenir une présentation par gène, par type de mutation (mutations activatrices, inactivatrices), par identité de la voie cellulaire dans laquelle il est impliqué, par mode de transmission de la prédisposition (dominant, récessif), par maladie prédisposante, par localisation tumorale ? Les connaissances récentes viennent encore compliquer toute tentative de présentation simple : en effet, les mêmes gènes peuvent conduire à des pathologies bien différentes selon que les altérations constitutionnelles sont mono- ou bi-alléliques. Il en est ainsi, notamment, des altérations de BRCA2, à l’origine d’un risque élevé de cancer du sein en cas d’altération mono-allélique, mais responsable d’une maladie de Fanconi en cas d’altération bi-allélique [10]. De la même façon, des altérations bi-alléliques des gènes hMSH2 et hMLH1 sont à l’origine de tumeurs cérébrales et d’hémopathies extrêmement évolutives, loin du tableau familial classique du syndrome HNPCC associé à une altération mono-allélique [11]. De plus, l’appartenance à la catégorie des gènes suppresseur de tumeur ou à celle des gènes de stabilité du génome n’est plus exclusive : à titre d’exemple, le gène APC est impliqué dans la voie Wnt de transduction du signal mitogène, mais aussi dans la ségrégation des chromosomes lors de la mitose [12]. Le Tableau I privilégie ainsi une présentation combinant l’appartenance à un groupe de gènes (suppresseur de tumeur, oncogène, gène impliqué dans la stabilité du génome) et l’identité de la voie cellulaire principale dans laquelle ils sont impliqués. On peut se référer pour une présentation plus clinique à l’article de revue récent de Garber et Offit [13].
Reprenant la métaphore de l’iceberg, il est clair que l’essentiel des facteurs de prédisposition aux cancers, la partie immergée, n’est pas identifié. Les dix ou vingt prochaines années du développement de l’oncogénétique seront celles de la compréhension des situations de prédispositions complexes, car multigéniques et faisant intervenir l’environnement (voir plus loin).
Diagnostic d’une prédisposition aux cancers
Les prédispositions aux cancers faisant l’objet de tests génétiques diagnostiques sont celles pour lesquelles les risques tumoraux ont été établis et la prise en charge des risques définie. Cette prise en charge, qui a pour finalité de diminuer la morbidité et la mortalité, est, dans la majorité des cas, une surveillance précoce et rapprochée conduite en milieu spécialisé. Cependant, dans certains cas, une prévention chirurgicale est recommandée : colectomie en cas de polypose adénomateuse clinique, ovariectomie dès l’âge de 40 ans en cas d’altération du gène BRCA1, par exemple. Par ailleurs, les efforts de recherche portent sur le développement de molécules ayant un effet préventif.
Les éléments évocateurs d’une prédisposition génétique sont le jeune âge au diagnostic (par rapport à l’âge moyen de l’atteinte dans la population), la multiplicité des lésions tumorales, la présence d’une histoire familiale du même cancer, ou d’autres cancers, associés à la prédisposition, et l’existence éventuelle d’une maladie prédisposante. Pour les cancers fréquents (sein, côlon), l’histoire familiale est un élément crucial : en général, est retenue comme évocatrice d’une prédisposition l’existence de trois cas unis entre eux par un lien de premier ou de second degré et appartenant à la même branche parentale. En raison de la grande diversité des mutations, qui sont différentes, en règle, d’une famille à l’autre, et surtout du fait qu’une mutation clairement délétère n’est pas toujours identifiée, les tests sont conduits en deux temps : analyse d’un cas index (en général une personne qui a été traitée pour un cancer donné à un âge précoce), puis le cas échéant, analyse des apparentés. De fait, si aucune mutation n’a été identifiée à l’issue de l’analyse du cas index, le test est considéré comme peu informatif, car on ne peut exclure l’existence d’une mutation délétère, et aucun test présymptomatique ne peut être proposé aux apparentés ; en revanche, si une mutation a été identifiée, un test de prédisposition peut leur être proposé : s’il est négatif, on peut retenir que l’apparenté testé n’a pas hérité du facteur familial de prédisposition.
Si la sélection d’une personne atteinte et ayant une histoire familiale permet de multiplier par 100 la probabilité d’identifier une mutation d’un gène BRCA1/2[1], ces critères de sélection ne sont cependant pas toujours satisfaisants. En effet, plus de 30 % des femmes atteintes de cancer du sein avant l’âge de 36 ans et porteuses d’une altération pourraient n’avoir aucune histoire familiale (D. Stoppa-Lyonnet, données non publiées), en raison de la pénétrance incomplète de la maladie (des femmes prédisposées n’auront pas développé de cancer du sein), d’une transmission paternelle, de la petite taille des familles ou encore, tout simplement, du fait de la méconnaissance de l’histoire familiale. Les analyses globales sur puces CGH (hybridation génomique comparative) et puces d’expression ont montré une certaine spécificité des cancers du sein BRCA1 et BRCA2 ; en d’autres termes, une signature génétique tumorale permet de s’affranchir de la sélection des cas prédisposés sur la seule histoire familiale. Dans le cadre des cancers du côlon, la recherche du phénotype tumoral MSI (voir plus haut) est déjà un élément d’orientation devant une histoire familiale atténuée de cancer du côlon.
Avec une fréquence de femmes prédisposées aux cancers du côlon et aux cancers du sein de l’ordre de 1/250 et d’hommes prédisposés aux cancers du côlon de l’ordre de 1/500 (Tableau I), les prédispositions aux cancers fréquents[2] obéissant à un modèle monogénique dominant font partie des maladies génétiques les plus fréquentes. Les consultations de génétique et les tests réalisés sont en plein essor : grâce à un programme de soutien de la DHOS (Direction de l’hospitalisation et de l’organisation des soins) relayée par l’INCa (Institut national du cancer), le nombre de tests de prédisposition aux cancers a été en 2003 de 11 659 et a progressé en 2004 de 24 % (14 452 tests) (communication C Bara, INCa). Ce programme s’est attaché à associer analyses de laboratoire, consultations de génétique et prise en charge multidisciplinaire des personnes à haut risque. Il favorise également une activité de réseaux entre les praticiens, afin d’homogénéiser les pratiques sur l’ensemble du territoire. Les mesures de prévention des cancers du sein et du côlon ont fait l’objet de consensus récemment publiés [15, 16]. En dehors de la définition des bonnes pratiques des tests, le réseau de laboratoires doit relever un défi : la classification des variants génétiques (mutations faux-sens, suspicion d’anomalie d’épissage) dont la signification biologique est inconnue. En effet, jusqu’à 40 % des mutations identifiées dans les gènes BRCA1 et BRCA2, de même que dans les gènes de prédisposition aux cancers du côlon, sont des variants de signification inconnue ; il est clair que la mise en commun par les laboratoires d’un certain nombre d’observations devrait permettre de mieux classer les patients et d’améliorer le prise en charge des familles.
Autres gènes de prédisposition aux cancers
Aujourd’hui, une grande partie des formes familiales de cancers fréquents (sein ou côlon) n’est pas expliquée par la ségrégation de l’allèle muté d’un gène déjà identifié. Les études d’épidémiologie génétique suggèrent l’existence de modes de transmission récessifs ou oligogéniques. L’identification des gènes responsables passe alors par des études d’association puissantes et/ou des études de liaison non paramétriques (paires de germains atteints), reposant sur la constitution de groupes de patients et de témoins judicieusement sélectionnés et nécessitant de très nombreux génotypages [17, 18]. De plus, il faut prendre en compte les interactions avec l’environnement, autre source de difficultés de ces analyses [19]. Enfin, on peut s’attendre à ce que ces gènes, une fois identifiés, soient chacun associés à une élévation modérée du risque tumoral ; l’histoire familiale sera alors rarement révélatrice d’une prédisposition. Les indications des tests seront tributaires des capacités d’analyse génétique et de la définition d’une prise en charge adaptée, une limite commune à toutes les maladies multifactorielles. Les polymorphismes des gènes du métabolisme des mutagènes et de certaines hormones, de la régulation de la réponse immune, de la réparation de l’ADN, voire de l’angiogenèse sont de très bon candidats [17], dont l’identification devrait ouvrir de nouvelles voies de recherche dans la chimioprévention des cancers.
Conclusions et perspectives
L’essentiel des gènes de prédisposition aux cancers obéissant à un modèle mendélien dominant ont été identifiés, d’autres associés à des prédispositions au déterminisme plus complexe devraient sans doute être identifiés dans les prochaines années. Néanmoins, il reste pour les premiers gènes identifiés encore bien des questions à résoudre. Pourquoi, alors qu’un gène a une expression ubiquitaire, son altération est-elle associée à un risque tumoral spécifique d’un tissu ? Il en est ainsi de la prédisposition au rétinoblastome : alors que la protéine RB a un rôle clé dans le contrôle du cycle cellulaire, son absence a un effet tumoral limité à la fois dans l’espace et dans le temps, les neuroblastes de la rétine de l’enfant de moins de deux ans principalement. Une autre question concerne les gènes dont l’altération transmise selon le mode dominant nécessite une inactivation somatique du second allèle. Existe-t-il une haplo-insuffisance dans les cellules non tumorales des personnes mutées ? La réponse à cette question est très importante pour les gènes de stabilité du génome : en effet, si l’altération d’un seul allèle BRCA1 ou BRCA2 est associée à un défaut de réparation de l’ADN, alors on peut craindre un effet mutagène plus important des rayons X utilisés pour la surveillance mammaire.
Enfin, nous n’avons évoqué dans cet article que les gènes associés à une augmentation du risque de survenue d’un cancer. Il faut s’interroger maintenant sur l’existence de déterminants génétiques constitutionnels influençant la réponse à la chimiothérapie et à la radiothérapie, mais aussi le processus métastatique. Quelle est la part des variations génétiques constitutionnelles ? En d’autres termes, les réponses aux traitements et le processus métastatique sont-ils inscrits dans le seul patrimoine génétique de la tumeur, ou également dans celui de l’hôte ? Il s’agit là d’un champ de la pharmacogénétique en pleine ouverture, l’identification de ces facteurs génétiques étant un grand défi. Les recherches reposeront en partie sur la constitution de banques de tumeurs ayant des annotations cliniques et associées à la conservation d’ADN constitutionnel. L’enjeu est de proposer un traitement individuel le plus adapté possible.
Appendices
Remerciements
Nous remercions Christine Bara, Anne-Paule Gimenez-Roqueplo, Pierre Laurent-Puig et Sylviane Olschwang pour les précisions qu’ils ont apportées au manuscrit ; Agnès Chompret, Isabelle Coupier, Marion Gauthier-Villars et Claude Houdayer pour leur relecture attentive du manuscrit. Nous remercions également les deux experts de médecine/sciences pour leurs suggestions constructives.
Notes
-
[1]
La fréquence des porteurs de mutations BRCA1 et BRCA2 dans la population est estimée à 1/500 [14]. Aujourd’hui, une mutation de l’un de ces gènes est identifiée dans environ 20 % (1/5) des cas index testés selon les critères fondés sur l’histoire familiale, évoqués plus haut. Ainsi, ces critères permettent de multiplier par 100 la probabilité de trouver une mutation (1/500 -> 1/5).
-
[2]
Prédisposition aux cancers du sein et du côlon. Les prédispositions au cancer de la prostate sont mises à part, pour l’instant, puisque le (ou les) gène(s) responsable(s) n’ont pas été définitivement identifiés.
Références
- 1. Vogelstein B, Kinzler KW. Cancer genes and the pathways they control.Nat Med 2004 ; 10 : 789-99.
- 2. Futreal PA, Coin L, Marshall M, et al. A census of human cancer genes.Nat Rev Cancer 2004 ; 4 : 177-83.
- 3. Hall JM, Lee MK, Newman B, et al. Linkage of early-onset familial breast cancer to chromosome 17q21.Science 1990 ; 250 : 1684-9.
- 4. Easton DF, Bishop DT, Ford D, et al. Genetic linkage analysis in familial breast and ovarian cancer : results from 214 families. The breast cancer linkage consortium.Am J Hum Genet 1993 ; 52 : 678-701.
- 5. Miki Y, Swensen J, Shattuck-Eidens D, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1.Science 1994 ; 266 : 66-71.
- 6. De la Chapelle A. Genetic predisposition to colorectal cancer.Nat Rev Cancer 2004 ; 4 : 769-80.
- 7. Pasca di Magliano M, Hebrok M. Hedgehog signalling in cancer formation and maintenance.Nat Rev Cancer 2003 ; 3 : 903-11.
- 8. Waite KA, Eng C. From developmental disorder to heritable cancer : it’s all in the BMP/TGF-beta family.Nat Rev Genet 2003 ; 4 : 763-73.
- 9. Eng C, Kiuru M, Fernandez MJ, et al. A role for mitochondrial enzymes in inherited neoplasia and beyond.Nat Rev Cancer 2003 ; 3 : 193-202.
- 10. Howlett NG, Taniguchi T, Olson S, et al. Biallelic inactivation of BRCA2 in Fanconi anemia.Science 2002 ; 297 : 606-9.
- 11. Bougeard G, Charbonnier F, Moerman A, et al. Early onset brain tumor and lymphoma in MSH2-deficient children.Am J Hum Genet 2003 ; 72 : 213-6.
- 12. Fodde R, Smits R, Clevers H. APC, signal transduction and genetic instability in colorectal cancer.Nat Rev Cancer 2001 ; 1 : 55-67.
- 13. Garber JE, Offit K. Hereditary cancer predisposition syndromes.J Clin Oncol 2005 ; 23 : 276-92.
- 14. Antoniou AC, Pharoah PD, McMullan G, et al. A comprehensive model for familial breast cancer incorporating BRCA1, BRCA2 and other genes.Br J Cancer 2002 ; 86 : 76-83.
- 15. Eisinger F, Bressac B, Castaigne D, et al. Identification et prise en charge des prédispositions héréditaires aux cancers du sein et de l’ovaire (mise à jour 2004).Bull Cancer 2004 ; 91 : 219-37.
- 16. Olschwang S, Bonaiti C, Feingold J, et al. Identification et prise en charge du syndrome HNPCC (hereditary non polyposis colon cancer), et des prédispositions héréditaires aux cancers du côlon et de l’endomètre.Bull Cancer 2004 ; 91 : 303-15.
- 17. Houlston RS, Peto J. The search for low-penetrance cancer susceptibility alleles.Oncogene 2004 ; 23 : 6471-6.
- 18. Pharoah PD, Dunning AM, Ponder BA, et al. Association studies for finding cancer-susceptibility genetic variants.Nat Rev Cancer 2004 ; 4 : 850-60.
- 19. Andrieu N, Goldstein AM. The case-combined-control design was efficient in detecting gene-environment interactions.J Clin Epidemiol 2004 ; 57 : 662-71.
List of tables
Tableau I
Gènes de prédisposition aux cancers identifiés.
Tableau I (continuation)
La fréquence des porteurs dans la population générale est donnée à titre indicatif et est susceptible de varier d’un facteur 2, voire plus pour les fréquences très faibles. Lorsque la fréquence est inconnue (notée ?), elle est a priori très faible. WAGR : Wilms tumor-aniridia-genitourinary anomalies-mental retardation ; HNPCC : hereditary non polyposis carcinoma ; HIF-1 : hypoxemia induced factor-1 ; PI3K : phosphatidylinositol 3 kinase ; RB : protéine du rétinoblastome ; RTK : récepteurs des tyrosine kinases ; HNF1 : hepatocyte nuclear factor 1 ; BER : base excision repair ; CIN : instabilité chromosomique ; MMR : mismatch repair ; NER : nucleotide excision repair (complété d’après [1]).