Abstracts
Résumé
Les histones sont les pièces maîtresses de la compaction de l’ADN en chromatine et jouent un rôle majeur dans la régulation des fonctions du génome. Elles sont les cibles de multiples modifications post-traductionnelles qui apportent une information épigénétique. L’ensemble de ces modifications constituerait un « code histone », permettant d’associer à chaque combinaison de modifications un état particulier de la chromatine. De surcroît, les histones se déclinent sous forme de variants dont on sait qu’ils diffèrent par leur séquence, leur fonction et leur mécanisme d’incorporation dans la chromatine. Ce répertoire élargi d’informations permet d’envisager de nouvelles possibilités de régulation épigénétique.
Summary
Histones are the fundamental structural proteins intimately associated with eukaryotic DNA to form a highly ordered and condensed nucleoproteic complex termed chromatin. They are the targets of various posttranslational modifications including acetylation, methylation, phosphorylation and ubiquitination that modulate the structure/function of chromatin. The combinatorial nature of histone modifications is hypothesized to define a « histone code » that considerably extends the information potential of the genetic code, giving rise to epigenetic information. Moreover, most core histones consist of several nonallelic variants that can mark specific loci and could play an important role in establishment and maintenance of epigenetic memory. Here we will briefly present our current knowledge about histone posttranslational modifications and their implications in the regulation of epigenetic information. We will next describe core histone variants, insisting on their mode of incorporation into chromatin to discuss their epigenetic function and inheritance.
Article body
Dans sa définition moderne, le terme épigénétique désigne des paramètres, héritables au cours des divisions cellulaires, qui contribuent à la régulation d’états fonctionnels au sein d’une cellule sans affecter directement la séquence d’ADN. Cette appréciation de l’importance de paramètres non codés génétiquement a pris un essor particulier avec les récents progrès réalisés dans la connaissance de l’organisation du génome au sein du noyau des cellules.
En effet, le génome nucléaire s’organise en une structure nucléoprotéique, appelée chromatine, qui, outre sa composante génétique, est riche d’une information épigénétique [1, 2]. Au niveau moléculaire, son unité élémentaire, le nucléosome, comprend une particule coeur et une région internucléosomique. La particule coeur est composée de 146 paires de bases d’ADN enroulées autour d’un octamère protéique comprenant deux copies de chacune des histones H2A, H2B, H3 et H4 (Figure 1A) [3], la région internucléosomique étant pour sa part caractérisée par la présence de l’histone H1 (histone « lien »). C’est donc la chromatine, et non l’ADN seul, qui est impliquée dans tous les événements moléculaires faisant intervenir le matériel génétique, à savoir la réplication, la transcription, la réparation et la recombinaison.
Figure 1
Particule coeur du nucléosome et modifications des histones.
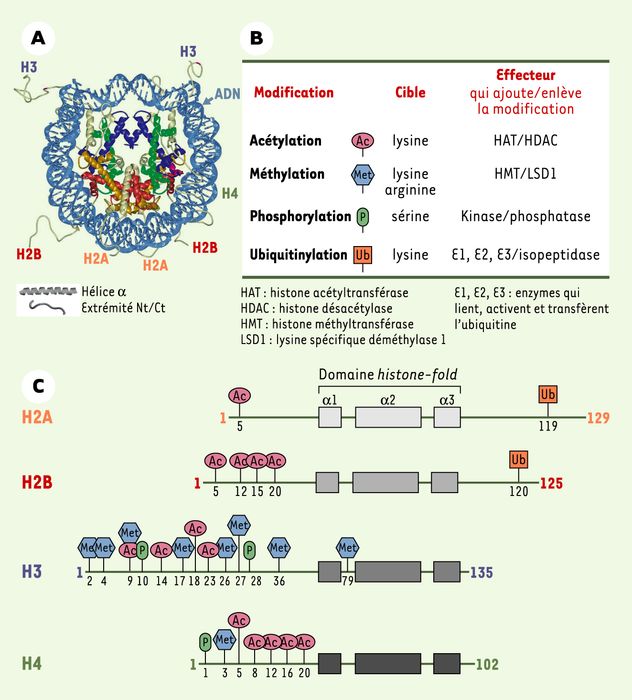
A. Structure de la particule coeur du nucléosome. La double hélice d’ADN (en bleu clair) s’enroule autour d’un octamère protéique constitué de deux molécules de chaque histone : H2A (en jaune), H2B (en rouge), H3 (en bleu) et H4 (en vert). Nt/Ct indique les extrémités amino- et carboxyterminales (d’après [3]). B. Les extrémités amino- et carboxyterminales des histones sont les cibles privilégiées de modifications post-traductionnelles : les mieux caractérisées sont l’acétylation, la méthylation, la phosphorylation et l’ubiquitinylation. Leurs cibles et leurs effecteurs (enzymes qui catalysent l’ajout ou la suppression de la modification) sont indiqués. Une seule histone déméthylase a été décrite à ce jour, LSD1 (lysine specific demethylase 1), qui est capable de déméthyler la lysine 4 de l’histone H3. C. Structure et modifications des histones majeures de la particule coeur du nucléosome chez l’homme. Les trois hélices α du motif conservé histone-fold, intervenant dans la dimérisation des histones, sont représentées par des rectangles gris. La nature et la position des modifications post-traductionnelles sont schématisées comme en B.
L’information épigénétique au sein de la chromatine est principalement véhiculée par des modifications de l’ADN et des histones. La modification majeure de l’ADN est la méthylation de la cytosine, qui est généralement la marque d’une chromatine transcriptionnellement silencieuse chez les vertébrés [4]. Quant aux histones, différentes modifications post-traductionnelles ont été décrites, comme l’acétylation, la phosphorylation, la méthylation et l’ubiquitinylation [5-7]. De plus, la nature même des histones, qui existent sous différentes formes appelées variants, est également une donnée épigénétique dont le rôle majeur a récemment été mis en avant [8]. Dans cet article, nous nous intéresserons aux principales modifications des histones de la particule coeur, en mettant tout particulièrement l’accent sur les nouvelles données concernant les variants d’histones et leur importance dans la modulation de l’information épigénétique.
Modifications post-traductionnelles des histones : le « code histone »
Les histones sont de petites protéines basiques (11-22 kDa) contenant un domaine globulaire de nature hydrophobe, le domaine histone-fold. Ce domaine, formé de trois hélices α, est impliqué dans la dimérisation des histones. De part et d’autre de ce domaine, très conservé, s’étendent les extrémités amino- et carboxyterminales qui émergent à la surface du nucléosome. Les extrémités aminoterminales, et dans une moindre mesure carboxyterminales, sont les cibles privilégiées de nombreuses modifications post-traductionnelles, bien que de nouvelles modifications aient également été identifiées dans d’autres régions. Les modifications les plus étudiées à ce jour sont l’acétylation, la méthylation, la phosphorylation et l’ubiquitinylation, mais d’autres modifications ont également été décrites telles que l’ADP-ribosylation, la sumoylation, la glycosylation ou la biotinylation. De plus, le nombre de modifications identifiées continue à s’accroître, grâce notamment à l’application de nouvelles approches technologiques telle que la spectrométrie de masse [9, 10].
Ces modifications covalentes sont ciblées sur des résidus spécifiques pour chaque histone et sont catalysées, généralement de manière réversible, par des enzymes également spécifiques (Figure 1B et C). Des familles d’enzymes qui ajoutent ou enlèvent une modification ont été caractérisées, mettant en évidence la réversibilité de telles modifications. Toutefois, jusqu’à très récemment, il n’était pas connu d’enzyme capable de déméthyler les histones. Ce n’est qu’à la fin de l’année 2004 qu’une protéine, LSD1 (lysine specific demethylase 1), ayant une activité déméthylase sur la lysine 4 de l’histone H3, a été décrite [11]. De plus, la déméthylation des arginines de l’histone H3 a également été mise en évidence, mais par un processus indirect faisant intervenir une étape de désimination qui convertit l’arginine en citrulline qui ne peut plus être méthylée [12, 13]. La réversibilité des modifications d’histone, tout comme celle de la modification par méthylation des CpG de l’ADN, met en évidence la plasticité de l’information épigénétique, à l’inverse du code génétique stable en dehors des événements de mutagenèse. Ainsi, les modifications des histones modulent la structure de la chromatine, permettant de contrôler les fonctions cellulaires liées à l’ADN, comme la transcription, en réponse à des changements physiologiques dans la cellule.
Il est proposé que l’ensemble des modifications des histones constituerait un code, appelé « code histone », qui permettrait d’associer à chaque combinaison de modifications un état particulier de la chromatine [5-7]. La transduction du code histone (Figure 2) implique la transformation de l’information codée par les modifications d’histones en une réponse contrôlant les fonctions cellulaires liées à l’ADN. Cette transduction peut être la conséquence directe d’un changement de structure de la chromatine, mais peut également faire intervenir un intermédiaire protéique, un transducteur, qui interagit spécifiquement avec l’histone modifiée. Ainsi, l’histone H3 est reconnue par des protéines à chromodomaine, telles que HP1 (heterochromatin protein 1), lorsque sa lysine 9 est méthylée. Les histones acétylées interagissent quant à elles spécifiquement avec des protéines à bromodomaine.
Figure 2
Transduction du « code histone ».
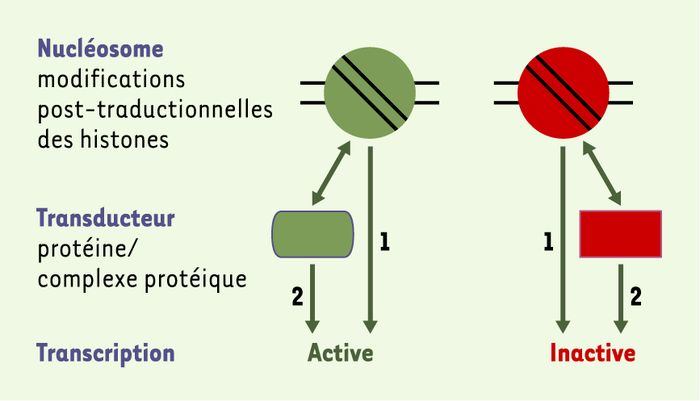
La combinaison entre les différentes modifications post-traductionnelles des histones présentes dans un nucléosome engendre, selon l’hypothèse du « code histone », un état actif ou inactif, notamment pour la transcription. Cet état peut-être la conséquence directe d’une structure particulière du nucléosome (1), ou résulter d’une interaction spécifique avec une protéine ou un complexe protéique (transducteur) (2).
La méthylation de la lysine 9 sur l’histone H3 constitue une marque - ou signature - associée aux régions du génome transcriptionnellement inactives, tandis que l’état hyperacétylé des histones correspond généralement aux régions actives. L’existence de plusieurs modifications sur une même histone apporte une dimension supplémentaire au code histone. En effet, une modification sur un site peut influencer la capacité d’un autre site à être modifié, de manière synergique ou au contraire antagoniste. Ainsi, la phosphorylation de la sérine 10 de l’histone H3 inhibe la méthylation de la lysine 9, mais est retrouvée associée aux lysines 9 et/ou 14 acétylées au cours d’une stimulation hormonale. Dans ce dernier cas, la phosphorylation/acétylation de l’histone H3 est la marque d’une région active transcriptionnellement, tandis que la phosphorylation de H3 est, dans un autre cas, corrélée à la condensation des chromosomes mitotiques. Le schéma actuel s’appuie donc sur une combinaison de modifications, et non une modification isolée, afin de conditionner un état particulier de la chromatine. Ce constat met en évidence le caractère très élaboré du code histone, dont le décryptage n’en est qu’à ses prémices.
En outre, ce code présente deux niveaux de lecture possibles. En toile de fond, les modifications stables au cours des divisions cellulaires, qu’on peut véritablement qualifier d’épigénétique selon la définition de Robin Holliday [14], telles que celles associées aux régions centromériques. Sur cette trame s’ajoutent les modifications qui permettent des changements d’états rapides et réversibles en réponse à des signaux externes, telles que la phosphorylation de l’histone H3 en réponse à un stress. Il est certainement intéressant, sur un plan évolutif, de réaliser que les mêmes types de modifications sont susceptibles d’être engagés dans deux modes d’utilisation : à court et à long terme (en transduction du signal ou pour une mémoire épigénétique).
Les variants d’histones : histones majeures et histones de remplacement
Chaque histone de la particule coeur, sauf H4, existe dans la cellule sous plusieurs formes protéiques, qui ont des homologies de séquences variables et sont codées par des gènes différents [15]. L’ensemble de ces formes pour chaque histone est regroupé sous le terme de « variants d’histones » (Figure 3A). Bien qu’ils soient connus depuis plusieurs dizaines d’années, les variants d’histones n’ont que très récemment fait l’objet d’un regain d’intérêt. Cet intérêt grandissant est en partie lié aux progrès technologiques qui permettent de localiser spécifiquement un variant d’histone, in vivo, en le fusionnant à la protéine fluorescente GFP (green fluorescent protein). Cette technique a permis de montrer, chez la drosophile, que le mode d’incorporation dans la chromatine du variant H3.3 diffère de celui du variant H3.2 [16].
Figure 3
Les variants d’histones.

Tableau récapitulatif (A) des variants des histones (majeures et de remplacement) de la particule coeur du nucléosome chez l’homme. Alignement des séquences protéiques des variants de l’histone H2A (B) et de l’histone H3 (C) chez l’homme (à partir du programme d’alignement de séquences : Clustal W). Les acides aminés en rouge (variants H2A) et en bleu (variants H3) correspondent à ceux qui sont différents de la séquence H2A.1 et H3.1, respectivement. Les pourcentages représentent l’identité de séquence en acides aminés avec H2A.1 ou H3.1. Les acides aminés différents entre H3.3 et H3.1 sont encadrés en rouge. Les positions des hélices α du motif histone-fold sont indiquées.
Schématiquement, il existe deux types de variants d’histones : les histones majeures et les histones de remplacement. On appelle histones majeures les histones dont l’expression est augmentée au cours de la phase S, afin de répondre au besoin massif en histones nécessaires à l’assemblage en chromatine de l’ADN en cours de réplication. Les histones de remplacement, minoritairement représentées, sont exprimées également en dehors de la phase S et peuvent être incorporées dans la chromatine indépendamment de la synthèse d’ADN [17]. Il est depuis peu apparu que certains variants sont associés à des structures ou à des fonctions spécifiques de la chromatine.
Deux variants de H2B, H2B.1 et H2B.2, sont des histones majeures, et un seul variant de remplacement, TSH2B (testis-sperm specific H2B), spécifique des tissus testiculaires et des spermatozoïdes, a été identifié à ce jour [18]. Les variants les mieux caractérisés sont ceux des histones H2A et H3. Six variants de l’histone H2A ont été décrits : deux histones majeures, H2A.1 et H2A.2, et quatre histones de remplacement, H2A.Z, H2A.X, macroH2A et H2A-Bbd (Barr body deficient) (Figure 3B) [19, 20]. Le variant H2A.Z joue un rôle essentiel dans le développement embryonnaire précoce chez les mammifères. Contrairement aux premières études, des données récentes ont montré qu’un nucléosome contenant H2A.Z serait plus stable que celui contenant l’une des histones majeures H2A. Les données concernant le rôle de H2A.Z sur l’activité transcriptionnelle sont également ambiguës : si certaines études démontrent que le variant H2A.Z semble important pour la formation de régions transcriptionnellement inactives, d’autres suggèrent que sa présence favoriserait la transcription. Le variant H2A.X est, quant à lui, phosphorylé en réponse aux cassures double-brin de l’ADN, et semble jouer un rôle important dans le processus de réparation de ces lésions. Le résidu cible de cette phosphorylation est la sérine 139, spécifique du variant H2A.X et située à son extrémité carboxyterminale. De son côté, le variant macroH2A est enrichi dans des régions silencieuses, notamment dans le chromosome X inactif (chez les femelles de mammifères), tandis que H2A-Bbd en est largement exclu, ce qui suggère pour ces deux variants des fonctions opposées dans la régulation de l’activité transcriptionnelle.
Concernant l’histone H3, deux histones majeures, H3.1 et H3.2, et deux histones de remplacement, H3.3 et CENP-A (centromeric protein A), ont été identifiées (Figure 3C). Le variant CENP-A est spécifiquement localisé dans les régions centromériques, où il semble essentiel à la structure et à la fonction des centromères, en participant directement à la formation d’un kinétochore actif [21]. Le variant H3.3 a, quant à lui, été retrouvé dans les régions d’ADN ribosomique transcriptionnellement actives chez la drosophile [16]. Dans des cellules humaines, ce variant s’accumule également dans les régions du génome dont la transcription a été activée, événement qui s’accompagne de la perte de la méthylation de H3 sur la lysine 9 (marque d’une région transcriptionnellement silencieuse) [22]. Aucune enzyme capable de déméthyler la lysine 9 de l’histone H3 n’ayant été découverte à ce jour, le remplacement d’une histone H3, dont la lysine 9 est méthylée, par le variant H3.3 pourrait être l’un des moyens utilisés par la cellule pour passer rapidement d’un état inactif à un état actif transcriptionnellement. De fait, une étude récente montre que le variant H3.3 est présent majoritairement sous une forme modifiée associée à un état transcriptionnel actif [23]. Toutefois, on ne sait pas encore si l’incorporation de H3.3 est réellement une cause, ou simplement une conséquence de l’activation de la transcription. De plus, H3.3 ne différant que par 4 et 5 acides aminés avec H3.2 et H3.1, respectivement, la manière dont un nucléosome contenant H3.3 favoriserait la transcription n’est pas encore comprise. Il faut également signaler qu’aucun anticorps ne permettant de différencier H3.1, H3.2 et H3.3, on ne peut aujourd’hui faire de distinction entre ces trois variants lors d’une étude sur les modifications des histones.
Enfin, des résultats récents montrent que les variants d’histones diffèrent également par leur mécanisme d’incorporation dans la chromatine.
Variants d’histones et modes d’incorporation dans la chromatine
La réplication de l’ADN au cours de la phase S du cycle cellulaire s’accompagne d’une duplication de son organisation en chromatine, ce qui permet la conservation de cette information épigénétique au fil des divisions cellulaires. L’assemblage des nucléosomes sur l’ADN en cours de synthèse est un processus qui se déroule schématiquement en deux étapes : l’incorporation d’un tétramère (H3-H4)2 dans l’ADN, puis l’ajout de deux dimères H2A-H2B [24]. Des protéines interagissant avec les histones, les chaperons d’histones, facilitent leur incorporation dans l’ADN. Au cours de la réplication, le tétramère (H3-H4)2 est déposé sur l’ADN grâce au chaperon CAF-1 (chromatin assembly factor 1), lui même ciblé sur l’ADN en cours de synthèse par la protéine PCNA (proliferating cell nuclear antigen) (Figure 4). L’incorporation des dimères H2A-H2B serait assurée par le chaperon d’histone NAP-1 (nucleosome assembly protein 1), ce qu’il reste toutefois à confirmer formellement. Si le mécanisme d’assemblage des nucléosomes au cours de la réplication est relativement bien établi, celui des histones de remplacement était inconnu jusqu’à très récemment.
Figure 4
Mécanismes d’incorporation des histones majeures et de remplacement dans l’ADN.
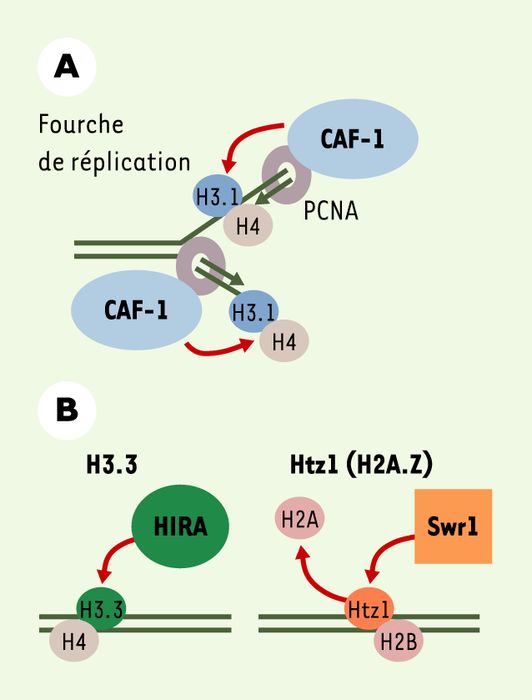
A. Les histones majeures sont assemblées en chromatine de manière couplée à la synthèse d’ADN (ici, la réplication). Les histones H3.1-H4 sont déposées sur l’ADN par le facteur d’assemblage CAF-1 (chromatin assembly factor 1), ciblé sur l’ADN en cours de synthèse par PCNA (proliferating cell nuclear antigen). B. Les histones de remplacement sont assemblées dans la chromatine par des mécanismes indépendants de la synthèse d’ADN. Le chaperon d’histones HIRA (histone regulating protein A) intervient dans l’incorporation du variant H3.3 dans l’ADN. Le complexe protéique contenant l’ATPase Swr1 (Swi2/Snf2 related) semble responsable de l’échange, chez la levure, de l’histone majeure H2A par l’histone de remplacement Htz1 (homologue de H2A.Z chez l’homme).
Puisque les histones de remplacement peuvent être incorporées dans la chromatine en dehors de tout événement de synthèse d’ADN, il est probable que la machinerie d’assemblage de ces histones est différente de celle utilisée par les histones majeures. De récents travaux réalisés chez la levure ont permis d’identifier un complexe capable de catalyser l’échange de l’histone majeure H2A par l’histone de remplacement Htz1 (homologue de H2A.Z chez l’homme). Une des sous-unités de ce complexe, Swr1 (Swi2/Snf2 related), est une adénosine triphosphatase (ATPase) de la famille des Swi2/Snf2 ATPases, constituants de facteurs de remodelage de la chromatine [25, 26]. Cette activité d’échange/incorporation d’histone représente un nouveau rôle pour ce type d’ATPases, seulement connues jusqu’à présent pour remodeler la structure de la chromatine sans en changer les constituants, et notamment les histones. De la même manière, la purification d’un complexe de pré-assemblage de l’histone de remplacement H3.3 a permis d’identifier la protéine chaperon impliquée dans sa mise en place sur l’ADN : il s’agit de la protéine HIRA (histone regulating protein A), capable d’assembler les nucléosomes indépendamment de la synthèse d’ADN [27, 28]. Dans ces mêmes études, le complexe associé à l’histone majeure H3.1 contenait le chaperon CAF-1, un facteur clé dans l’assemblage en chromatine couplé à la synthèse d’ADN, ce qui est en accord avec le mode d’incorporation observé pour les histones majeures.
Parmi les histones de remplacement, H2A.Z et H3.3 sont les seules pour lesquelles le mode d’incorporation dans la chromatine est en partie élucidé (Figure 4). À la lumière de ces résultats, il semble possible qu’à chaque histone de remplacement corresponde un mécanisme d’assemblage particulier faisant intervenir un complexe protéique spécifique. Il reste à comprendre, notamment, comment ces différents complexes d’assemblage peuvent être ciblés dans des régions précises du génome.
Les variants d’histones sont, au même titre que les modifications post-traductionnelles des histones, porteurs d’une information épigénétique. Ils contribuent à la variété et à la spécialisation des nucléosomes, définissant ainsi des domaines structuraux et fonctionnels dans le génome.
Conclusions
Une question d’actualité est de comprendre comment l’information épigénétique est maintenue au cours des divisions cellulaires. Cela est d’autant plus crucial qu’un grand nombre de dysfonctionnements cellulaires ne sont pas uniquement dus à des événements de mutation au niveau de la séquence d’ADN : des anomalies affectant des facteurs clés dans la mise en place de modifications des histones et dans l’organisation de domaines de chromatine sont en effet fréquemment associées à différentes maladies, et notamment au cancer [29, 30] ((→) m/s 2005, n° 4, p. 405). Le décodage de l’information portée par les histones, ainsi que l’étude du ciblage moléculaire de tels paramètres épigénétiques, représentent donc des pistes intéressantes à explorer, sur les plans diagnostique comme thérapeutique.
Appendices
Remerciements
Les travaux de notre équipe sont financés par le CNRS, l’Institut Curie, la Ligue Nationale contre le Cancer (Équipe labellisée la Ligue), le Programme collaboratif Institut Curie/CEA et le réseau d’excellence Épigénome.
Références
- 1. Kornberg RD. Structure of chromatin.Annu Rev Biochem 1977 ; 46 : 931-54.
- 2. Wolffe A. Chromatin : structure and function, 3rd ed. London-San Diego, California : Academic Press, 1998.
- 3. Luger K. Structure and dynamic behavior of nucleosomes.Curr Opin Genet Dev 2003 ; 13 : 127-35.
- 4. Bird A. DNA methylation patterns and epigenetic memory.Genes Dev 2002 ; 16 : 6-21.
- 5. Jenuwein T, Allis CD. Translating the histone code.Science 2001 ; 293 : 1074-80.
- 6. Turner BM. Histone acetylation and an epigenetic code.BioEssays 2000 ; 22 : 836-45.
- 7. Turner BM. Cellular memory and the histone code.Cell 2002 ; 111 : 285-91.
- 8. Henikoff S, Furuyama T, Ahmad K. Histone variants, nucleosome assembly and epigenetic inheritance.Trends Genet 2004 ; 20 : 320-6.
- 9. Bonaldi T, Imhof A, Regula JT. A combination of different mass spectroscopic techniques for the analysis of dynamic changes of histone modifications.Proteomics 2004 ; 4 : 1382-96.
- 10. Freitas MA, Sklenar AR, Parthun MR. Application of mass spectrometry to the identification and quantification of histone post-translational modifications.J Cell Biochem 2004 ; 92 : 691-700.
- 11. Shi Y, Lan F, Matson C, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1.Cell 2004 ; 119 : 941-53.
- 12. Wang Y, Wysocka J, Sayegh J, et al. Human PAD4 regulates histone arginine methylation levels via demethylimination.Science 2004 ; 306 : 279-83.
- 13. Cuthbert GL, Daujat S, Snowden AW, et al. Histone deimination antagonizes arginine methylation.Cell 2004 ; 118 : 545-53.
- 14. Holliday R. Epigenetics comes of age in the twentyfirst century.J Genet 2002 ; 81 : 1-4.
- 15. Franklin SG, Zweidler A. Non-allelic variants of histones 2a, 2b and 3 in mammals.Nature 1977 ; 266 : 273-5.
- 16. Ahmad K, Henikoff S. The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly.Mol Cell 2002 ; 9 : 1191-200.
- 17. Wu RS, Tsai S, Bonner WM. Patterns of histone variant synthesis can distinguish G0 from G1 cells.Cell 1982 ; 31 : 367-74.
- 18. Zalensky AO, Siino JS, Gineitis AA, et al. Human testis/sperm-specific histone H2B (hTSH2B). Molecular cloning and characterization.J Biol Chem 2002 ; 277 : 43474-80.
- 19. Chadwick BP, Willard HF. A novel chromatin protein, distantly related to histone H2A, is largely excluded from the inactive X chromosome.J Cell Biol 2001 ; 152 : 375-84.
- 20. Perche PY, Robert-Nicoud M, Khochbin S, Vourc’h C. Différenciation du nucléosome : le rôle des variants de l’histone H2A. Med Sci (Paris) 2003 ; 19 : 1137-45.
- 21. Sullivan KF. A solid foundation : functional specialization of centromeric chromatin.Curr Opin Genet Dev 2001 ; 11 : 182-8.
- 22. Janicki SM, Tsukamoto T, Salghetti SE, et al. From silencing to gene expression : real-time analysis in single cells.Cell 2004 ; 116 : 683-98.
- 23. McKittrick E, Gafken PR, Ahmad K, Henikoff S. Histone H3.3 is enriched in covalent modifications associated with active chromatin.Proc Natl Acad Sci USA 2004 ; 101 : 1525-30.
- 24. Kaufman PD, Almouzni G. DNA replication, nucleotide excision repair and nucleosome assembly. In : Elgin SCR, Workman JL, eds. Chromatin structure and gene expression. New York : Oxford University Press, 2000.
- 25. Krogan NJ, Keogh MC, Datta N, et al. A Snf2 family ATPase complex required for recruitment of the histone H2A variant Htz1.Mol Cell 2003 ; 12 : 1565-76.
- 26. Mizuguchi G, Shen X, Landry J, et al. ATP-driven exchange of histone H2AZ variant catalyzed by SWR1 chromatin remodeling complex.Science 2004 ; 303 : 343-8.
- 27. Ray-Gallet D, Quivy JP, Scamps C, et al. HIRA is critical for a nucleosome assembly pathway independent of DNA synthesis.Mol Cell 2002 ; 9 : 1091-100.
- 28. Tagami H, Ray-Gallet D, Almouzni G, Nakatani Y. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis.Cell 2004 ; 116 : 51-61.
- 29. Hake SB, Xiao A, Allis CD. Linking the epigenetic language of covalent histone modifications to cancer.Br J Cancer 2004 ; 90 : 761-9.
- 30. Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy.Nature 2004 ; 429 : 457-63.
List of figures
Figure 1
Particule coeur du nucléosome et modifications des histones.
A. Structure de la particule coeur du nucléosome. La double hélice d’ADN (en bleu clair) s’enroule autour d’un octamère protéique constitué de deux molécules de chaque histone : H2A (en jaune), H2B (en rouge), H3 (en bleu) et H4 (en vert). Nt/Ct indique les extrémités amino- et carboxyterminales (d’après [3]). B. Les extrémités amino- et carboxyterminales des histones sont les cibles privilégiées de modifications post-traductionnelles : les mieux caractérisées sont l’acétylation, la méthylation, la phosphorylation et l’ubiquitinylation. Leurs cibles et leurs effecteurs (enzymes qui catalysent l’ajout ou la suppression de la modification) sont indiqués. Une seule histone déméthylase a été décrite à ce jour, LSD1 (lysine specific demethylase 1), qui est capable de déméthyler la lysine 4 de l’histone H3. C. Structure et modifications des histones majeures de la particule coeur du nucléosome chez l’homme. Les trois hélices α du motif conservé histone-fold, intervenant dans la dimérisation des histones, sont représentées par des rectangles gris. La nature et la position des modifications post-traductionnelles sont schématisées comme en B.
Figure 2
Transduction du « code histone ».
La combinaison entre les différentes modifications post-traductionnelles des histones présentes dans un nucléosome engendre, selon l’hypothèse du « code histone », un état actif ou inactif, notamment pour la transcription. Cet état peut-être la conséquence directe d’une structure particulière du nucléosome (1), ou résulter d’une interaction spécifique avec une protéine ou un complexe protéique (transducteur) (2).
Figure 3
Les variants d’histones.
Tableau récapitulatif (A) des variants des histones (majeures et de remplacement) de la particule coeur du nucléosome chez l’homme. Alignement des séquences protéiques des variants de l’histone H2A (B) et de l’histone H3 (C) chez l’homme (à partir du programme d’alignement de séquences : Clustal W). Les acides aminés en rouge (variants H2A) et en bleu (variants H3) correspondent à ceux qui sont différents de la séquence H2A.1 et H3.1, respectivement. Les pourcentages représentent l’identité de séquence en acides aminés avec H2A.1 ou H3.1. Les acides aminés différents entre H3.3 et H3.1 sont encadrés en rouge. Les positions des hélices α du motif histone-fold sont indiquées.
Figure 4
Mécanismes d’incorporation des histones majeures et de remplacement dans l’ADN.
A. Les histones majeures sont assemblées en chromatine de manière couplée à la synthèse d’ADN (ici, la réplication). Les histones H3.1-H4 sont déposées sur l’ADN par le facteur d’assemblage CAF-1 (chromatin assembly factor 1), ciblé sur l’ADN en cours de synthèse par PCNA (proliferating cell nuclear antigen). B. Les histones de remplacement sont assemblées dans la chromatine par des mécanismes indépendants de la synthèse d’ADN. Le chaperon d’histones HIRA (histone regulating protein A) intervient dans l’incorporation du variant H3.3 dans l’ADN. Le complexe protéique contenant l’ATPase Swr1 (Swi2/Snf2 related) semble responsable de l’échange, chez la levure, de l’histone majeure H2A par l’histone de remplacement Htz1 (homologue de H2A.Z chez l’homme).