Abstracts
Résumé
En 2002, il était prévu que les anticorps humanisés deviendraient une classe majeure de médicaments, notamment en cancérologie. Aujourd’hui, huit d’entre eux sont utilisés en clinique et plus de quarante font l’objet d’essais cliniques. Si leurs mécanismes d’action, multiples, sont difficiles à préciser pour un anticorps donné, les observations réalisées chez les milliers de patients déjà traités démontrent clairement leur intérêt clinique. Ils sont cependant d’utilisation délicate, en particulier lorsqu’ils modulent l’activité d’effecteurs de la réponse immune. Au cours des prochaines années, de nouveaux progrès devront être faits pour sélectionner les cibles pertinentes de ces anticorps, diminuer leur immunogénicité et réduire leur coût.
Summary
Since 1997, nine humanized antibodies received the approval of the FDA to be used as drugs for the treatment of various diseases including transplant rejections, metastatic breast and colon cancers, leukaemia, non-Hodgkin lymphomas, allergic conditions or multiple sclerosis. This review describes techniques used to engineer these antibodies and presents the recent evolutions of these techniques : SDRs grafting or « abbreviated » CDRs grafting. Based on the illustrative examples of several antibodies, Mylotarg®, Herceptin® or Xolair®, the therapeutic effectiveness of humanized antibodies are underlined and, with the example of Tysabri®, the sometimes dramatic adverse effects associated with their clinical use is stressed. In a second part, this review presents some future and realistic avenues to improve the effectiveness of the humanized antibodies, to decrease their immunogenicity and to reduce their cost.
Article body
Dès le début du siècle dernier, un immunologiste, Paul Ehrlich, eut la vision que les anticorps seraient utilisés en thérapeutique comme des « balles magiques » [1]. Il a fallu près d’un siècle et la découverte des anticorps monoclonaux en 1975 par Georges Kohler et Cesar Milstein pour que cette vision se transforme en réalité [2]. Cette découverte permettait enfin d’obtenir un anticorps de spécificité unique en quantité importante, et l’équipe de Ron Levy décrivait en 1982 le premier succès de l’utilisation d’un anticorps monoclonal (AcM) en thérapeutique [3]. Celui-ci, utilisé pour le traitement de lymphomes B, laissait entrevoir une large utilisation thérapeutique des anticorps monoclonaux. Pourtant, au cours des 12 années suivantes, un seul AcM, l’anticorps Orthoclone OKT®3 (muromonab-CD3), recevra une autorisation des autorités réglementaires américaines (Food and drug administration, FDA) pour une utilisation clinique, en l’occurrence le traitement du rejet aigu d’allogreffes rénales, hépatiques ou cardiaques. Il faudra attendre 1994 pour qu’un autre AcM, le ReoPro® (abxisimab), reçoive l’autorisation de la FDA pour une seconde utilisation clinique. Cet anticorps, dirigé contre un récepteur présent sur les plaquettes, est utilisé pour éviter la formation de caillots chez les patients ayant bénéficié d’une chirurgie cardiovasculaire.
En fait, les années 80 et la première moitié des années 90 ont été marquées par deux faits majeurs : le premier est l’échec de nombreux essais cliniques réalisés avec des anticorps monoclonaux, en particulier pour le traitement des cancers. Ces échecs répétés avaient pour principale cause l’origine murine des anticorps utilisés, qui induisent constamment la formation d’anticorps humains dirigés contre les anticorps murins (AHAM). Dans 84 % des cas, cette réponse immune est importante [4] (Figure 1), d’autant plus que l’anticorps est utilisé de façon répétée et à forte dose. Ces AHAM entraînent une élimination rapide des anticorps murins et des effets adverses parfois fatals. De plus, les anticorps murins ont une demi-vie courte dans le sérum, ainsi qu’une capacité limitée pour recruter des effecteurs cellulaires ou les protéines impliquées dans la réponse immune et réaliser une cytotoxicité cellulaire dépendante de l’anticorps ou une cytotoxicité dépendante du complément [5].
Figure 1
Pourcentage d'anticorps induisant une réponse immune anti-anticorps.
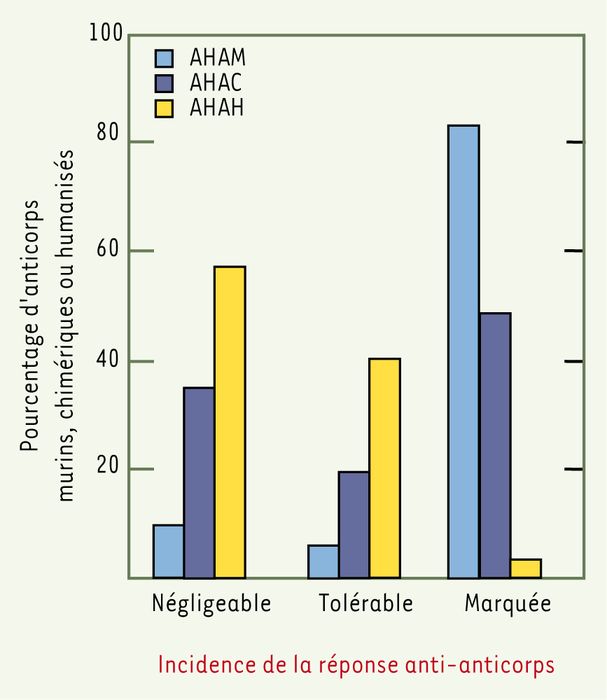
Une réponse est qualifiée de négligeable lorsqu'elle se produit chez moins de 2 % des patients, tolérable lorsqu'elle se produit chez 2 % à 15% des patients et marquée lorsqu'elle se produit chez plus de 15% d'entre eux. AHAM : anticorps humains anti-anticorps murins ; AHAC : anticorps humains anti-anticorps chimériques ; AHAH : anticorps humains anti-anticorps humanisés (d'après [4]).
Le second fait majeur est le développement de l’ingénierie génétique des anticorps, qui a permis de transformer progressivement les anticorps murins en anticorps humains. Cette transformation a d’abord conduit à la construction d’anticorps chimériques, dans lesquels les régions constantes des chaînes lourdes et légères des immunoglobulines murines sont remplacées par des régions constantes humaines [6] (Figure 2). Cette chimérisation conduit à une diminution importante des réponses immunes dirigées contre les régions murines (Figure 1) et a conduit au succès du ReoPro®, premier anticorps chimérique utilisé en clinique.
Figure 2
Formes d’anticorps monoclonaux utilisés en thérapeutique.

Anticorps chimérique : les régions constantes des chaînes lourdes et légères des immunoglobulines murines sont remplacées par des régions constantes humaines. Anticorps humanisé : les régions hypervariables (complementary determining region, CDR) d’un anticorps humain sont remplacées par des régions CDR d’origine murine, qui constituent le site de liaison de l’antigène. Cependant, certains acides aminés situés dans les régions adjacentes aux régions CDR (régions charpentes, ou FR pour framework) jouent un rôle majeur dans la structure du site de liaison de l’antigène. Pour conserver cette structure au cours de l’humanisation, l’anticorps accepteur reçoit non seulement les régions CDR de l’anticorps murin donneur, mais aussi les acides aminés de la région charpente de l’anticorps donneur.
Ce succès a été suivi rapidement par l’utilisation en clinique de trois autres anticorps chimériques : le Rituxan® (rituximab), en 1997, pour le traitement des lymphomes non hodgkiniens, le Simulect® (basiliximab), pour la prévention des rejets de greffe, et le Remicade® (infliximab), pour le traitement de la maladie de Crohn, en 1998. Cependant, la formation d’anticorps humains dirigés contre les anticorps chimériques (AHAC) est retrouvée chez 40 % des patients [4] (Figure 1), ce qui n’est pas surprenant puisque environ 30 % d’un anticorps chimérique est d’origine murine. Il est donc apparu nécessaire de rendre les anticorps murins encore plus humains.
Une humanisation des anticorps a été décrite pour la première fois par l’équipe de Greg Winter en 1986 [7]. Initialement, elle consiste à remplacer les régions hypervariables (complementary determining region, CDR) d’un anticorps humain par des régions CDR d’origine murine : l’anticorps ainsi construit est dit « humanisé », puisque moins de 10 % de ses séquences peptidiques sont d’origine murine.
Ingénierie des anticorps humanisés
En 1997, le Zenapax® (daclizumab) a été le premier anticorps humanisé utilisé en clinique (Tableau I), et la dernière décennie a été marquée par une utilisation thérapeutique de plus en plus fréquente de ce type d’anticorps. Le Zenapax® est la forme IgG1 humanisée de l’anticorps murin anti-Tac, dirigé contre la chaîne α du CD25, récepteur humain de l’interleukine-2 (IL-2) : cet anticorps bloque l’interaction de l’IL-2 avec son récepteur et prévient l’activation des cellules T. Approuvé en 1997 pour la prévention des rejets de greffe rénale, il a été administré à plus de 20 000 patients ; son efficacité et sa sûreté ont fourni la preuve de la validité du concept d’anticorps humanisé. Il est donc intéressant d’utiliser le Zenapax® comme exemple démonstratif d’une procédure d’humanisation d’un anticorps murin.
Tableau I
Anticorps monoclonaux humanisés utilisés en clinique.
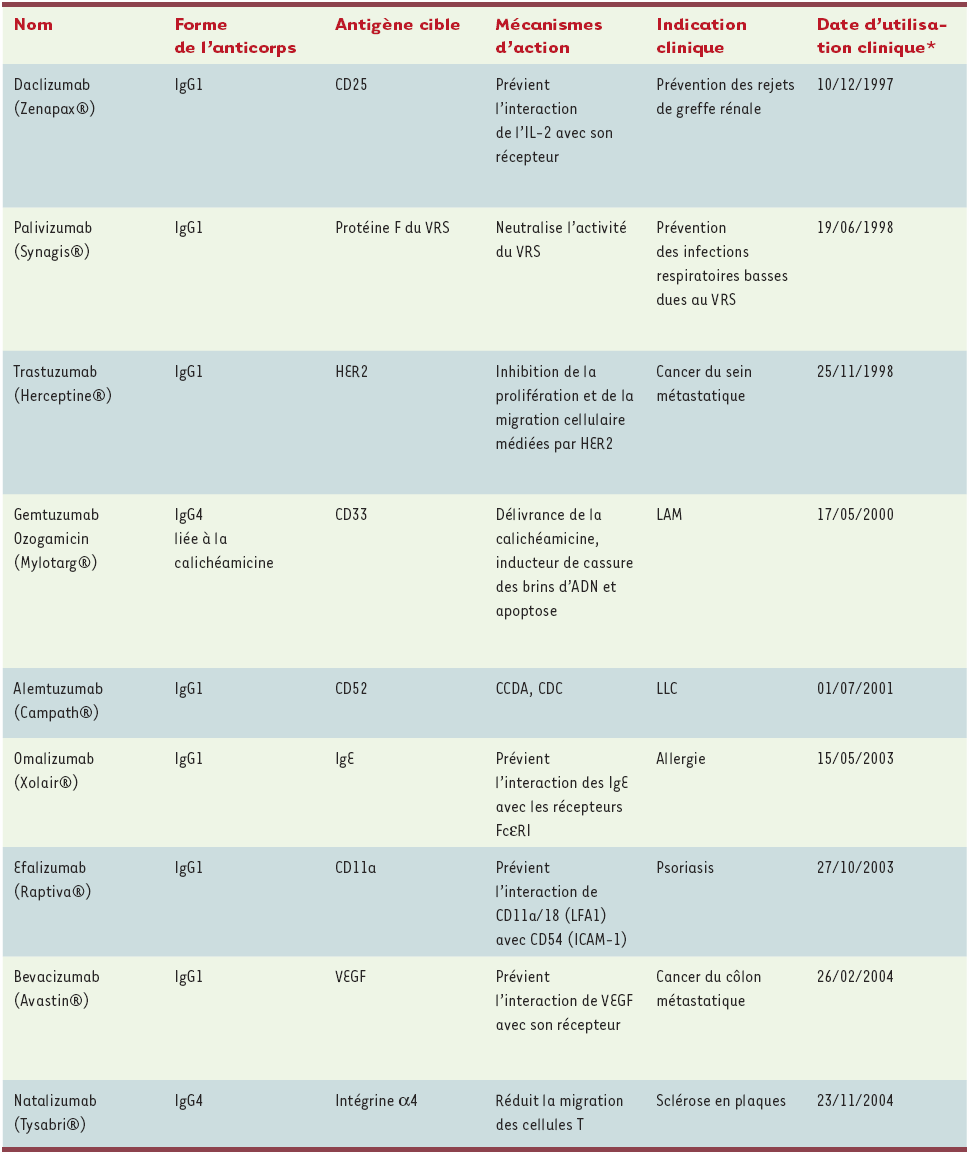
VRS : virus respiratoire syncitial ; HER2 : récepteur Erb2 du facteur de croissance épidermique humain ; VEGF : vascular endothelium growth factor ; IL-2 : interleukine-2 ; CCDA : cytotoxicité cellulaire dépendante de l’anticorps ; CDC : cytotoxicité dépendante du complément ; LNH : lymphome non hodgkinien ; LAM : leucémie aiguë myéloïde ; LLC : leucémie lymphoïde chronique. *Date d’approbation par les autorités américaines (Food and drug administration, FDA).
Principales étapes
Le but ultime de l’humanisation d’un anticorps est de produire des régions variables proches des régions humaines sans que l’anticorps humanisé perde l’affinité et la spécificité de l’anticorps murin. Pour atteindre ce but, il faut transférer dans un cadre humain « accepteur » des acides aminés des régions CDR provenant de l’anticorps monoclonal « donneur » d’origine murine (Figure 3). Ce concept repose sur le fait que les six régions CDR des régions variables des chaînes lourdes et légères contiennent la majorité des acides aminés constituant le site de liaison de l’antigène. En greffant les CDR provenant d’un anticorps monoclonal murin (l’anticorps « donneur ») dans des régions variables humaines (l’anticorps « accepteur »), on attend que les CDR murins puissent remplacer les CDR humains sans affecter la structure du site de liaison de l’antigène formé par les CDR murins. Si cela s’est avéré exact dans certains cas [8], la plupart des anticorps humanisés réalisés par greffage des régions CDR n’ont plus la même affinité pour l’antigène que l’anticorps murin, en raison du rôle majeur de certains acides aminés des régions adjacentes aux CDR (régions charpentes, ou FR pour framework) dans la structure du site de liaison de l’antigène. Pour conserver cette structure, il est donc nécessaire de greffer dans l’anticorps accepteur non seulement les régions CDR de l’anticorps murin donneur, mais aussi les acides aminés de la région charpente de l’anticorps donneur. Cela a été réalisé pour le Zenapax® et a nécessité plusieurs étapes.
Figure 3
Protocoles d’immunisation de la région variable d’une chaîne légère.

CDR : région déterminante complémentaire ; SDR : résidu déterminant spécifique (acide aminé des régions CDR critique dans l’interaction antigène-anticorps) : CDR abrégés : portions des CDR contenant les résidus SDR (d’après [9]).
La première étape consiste à cloner et à séquencer les gènes des régions variables de l’anticorps murin. Auparavant, il faut s’assurer que l’hybridome a bien été sous-cloné, grâce à un trieur de cellules ou par dilution limite. Le clonage est réalisé par transcription inverse et réaction d’amplification en chaîne (RT-PCR) avec des amorces spécifiques pour les gènes codant les régions variables des immunoglobulines murines. Il peut également être réalisé par technique rapide d’amplification des extrémités 5’ des ADNc (5’-RACE). Après RT-PCR ou 5’-RACE, les fragments d’ADN contenant les gènes codant pour les régions variables des chaînes lourdes (VH) et légères (VL) des immunoglobulines murines sont séquencés et les séquences des acides aminés des chaînes VH et VL sont déterminées.
La seconde étape, plus complexe, consiste à réaliser la conception (le design) de l’anticorps humanisé : il faut notamment identifier les résidus des régions charpentes murines importants pour maintenir la structure des CDR murins, afin de les transférer également dans un cadre humain. Ce dernier est lui-même sélectionné sur la base de son homologie avec les régions variables murines : cette étape commence par la réalisation d’un modèle en 3-dimensions (3-D) des régions variables grâce à des programmes informatiques. En parallèle, les régions variables VH et VL murines sont comparées avec les séquences de régions humaines présentes dans les bases de données, afin de choisir le cadre humain le plus approprié à l’humanisation. Pour l’anticorps murin anti-Tac, c’est l’anticorps humain Eu qui a été sélectionné : 67 % des acides aminés des régions charpentes des chaînes lourdes et 65 % des acides aminés des régions charpentes des chaînes légères étaient identiques entre l’anticorps murin anti-Tac et l’anticorps humain Eu. Cet anticorps est, avec les anticorps NEWM, KOL et REI, parmi les plus utilisés comme cadre « accepteur ».
Lorsque le modèle 3-D des régions variables murines et le choix du cadre humain sont effectués, il faut alors identifier les résidus des régions charpentes susceptibles de contribuer au site de liaison de l’antigène et ceux qui peuvent représenter une source potentielle d’immunogénicité. Il est également prudent d’examiner les séquences pouvant être des sites de liaisons de chaînes glucidiques. Enfin, il faut choisir l’isotype de l’anticorps humain. Des programmes informatiques tels que RASMOL sont utilisés pour rechercher les résidus des régions charpentes susceptibles de participer au site de liaison de l’antigène : ils identifient notamment les acides aminés à une distance d’environ 4 à 6 Å des régions CDR. Si les résidus sélectionnés diffèrent entre les régions charpentes murines et humaines, il faut alors considérer l’inclusion d’acides aminés murins dans l’anticorps humanisé. Or, certains de ces acides aminés sont potentiellement immunogéniques : il faut alors rechercher, dans les régions charpentes murines, les résidus « atypiques », c’est-à-dire rarement retrouvés dans les régions charpentes humaines. Ces résidus sont alors remplacés dans le cadre humain par le ou les résidus les plus fréquemment retrouvés dans les régions charpentes humaines aux positions correspondantes. Pour éliminer les sites potentiels de N-glycosylation, on effectue des changements d’acides aminés susceptibles de ne modifier que légèrement la conformation des régions CDR. Enfin, le choix de l’isotype de l’immunoglobuline s’effectue souvent en faveur de l’isotype IgG1, sauf s’il faut minimiser les mécanismes effecteurs de l’anticorps pour éviter certains effets adverses.
La troisième étape consiste à produire les anticorps humanisés. Après synthèse des régions variables selon la conception choisie, souvent par amplification PCR de séquences chevauchantes, il faut choisir les vecteurs d’expression stable dans les cellules telles que la lignée d’hybridome de souris Sp2/0-Ag14, la lignée CHO ou la lignée NSO.
Après expression de l’anticorps humanisé, les dernières étapes consistent à vérifier l’affinité et l’activité biologique de l’anticorps. Une description détaillée de l’ensemble de ces étapes peut être trouvée dans des revues récentes [5, 8].
Évolutions récentes
Depuis la conception et la construction des premiers anticorps humanisés, des améliorations constantes tentent d’être apportées dans l’ingénierie des anticorps humanisés. Par exemple, la technique de greffage de résidus déterminants spécifiques (specificity determining residues, SDR) repose sur l’analyse de multiples structures, en 3-D, des sites de liaison de l’antigène. Cette analyse suggère que seuls 20 % à 33 % des acides aminés des régions CDR sont critiques dans l’interaction antigène-anticorps. Ces résidus, les SDR, sont uniques pour chaque anticorps. Une nouvelle approche de l’humanisation est donc fondée sur le greffage non plus de l’ensemble des régions CDR, mais seulement des SDR de l’anticorps murin dans les régions variables humaines [9] (Figure 3). Cette approche de l’humanisation, sensée diminuer l’immunogénicité potentielle tout en optimisant l’affinité de l’anticorps, nécessite l’identification des SDR. Si cette identification peut être réalisée par détermination de la structure 3-D du complexe antigène-anticorps, elle n’est effective que pour un nombre limité d’anticorps. En l’absence de structure 3-D, il faudra rechercher, à l’aide des bases de données, les séquences variables humaines les plus proches des séquences murines : les différences d’acides aminés entre les séquences murines des régions VL et VH et les séquences humaines les plus similaires sont alors identifiées. En s’aidant de structures connues de complexes antigène-anticorps, on tentera d’identifier, parmi les acides aminés caractéristiques des régions variables murines, ceux localisés à des positions qui les mettent en contact direct avec l’antigène : les résidus murins localisés dans ces positions de contact sont très probablement des SDR, et devront être inclus dans le cadre humain lors de l’humanisation. Cependant, la prédiction des SDR par l’informatique reste difficile. Un autre moyen de choisir les SDR consiste à réaliser de nombreux mutants de l’anticorps murin en substituant chaque acide aminé murin susceptible d’être impliqué dans le site de liaison par l’acide aminé du cadre humain : si la mutation n’affecte pas l’affinité de l’anticorps mutant, l’acide aminé humain sera utilisé. À l’inverse, une mutation modifiant fortement l’affinité suggère que l’acide aminé murin est un SDR devant être conservé lors de l’humanisation.
Une autre approche de l’humanisation consiste à greffer des régions « CDR abrégés », les limites des régions CDR étant en partie connues : par exemple, la région CDR1 des chaînes légères s’étend des résidus 24 à 34. Comme les SDR situés à l’intérieur des CDR sont les résidus importants, les portions de CDR les contenant devraient être suffisantes pour l’humanisation. Toujours pour exemple, cette portion de la région CDR1 des chaînes légères inclut les résidus 27 à 34 ; ces CDR abrégés peuvent être utilisés à la place des CDR pour l’humanisation des anticorps (Figure 3).
Les évolutions techniques présentées dans cette revue ne sont pas exhaustives, et d’autres approches, telle l’utilisation de lignées germinales comme cadre humain accepteur, sont proposées. De même, la technique de variable domain resurfacing est toujours proposée comme une alternative à la technique de greffage des régions CDR [10] : cette technique, qui consiste à remplacer les résidus exposés à la surface dans les régions charpentes des anticorps murins par les résidus habituellement trouvés à la surface des anticorps humains [11], est fondée sur la notion que les anticorps humains dirigés contre les régions variables des anticorps murins sont dirigés contre les résidus murins localisés à la surface [12]. Si les techniques de biologie moléculaire utilisées pour réaliser cette approche sont classiques, la conception de l’anticorps à humaniser par variable domain resurfacing n’est, toutefois, pas toujours aisée, et l’influence des modifications effectuées sur l’affinité des anticorps pas toujours maîtrisée.
Anticorps humanisés utilisés en clinique
En janvier 2005, neuf anticorps humanisés avaient reçu l’aval de la FDA pour être utilisés en clinique. Au cours des sept dernières années, des milliers de malades ont donc pu bénéficier de ce type de traitement. Si les observations effectuées au cours de ces années ont confirmé l’efficacité de ces anticorps en clinique, elles ont aussi montré la difficulté de leur utilisation, avec des effets adverses imprévisibles et un coût élevé (Tableau I).
Une efficacité remarquable
Dès 1997, il a été démontré que les anticorps humains avaient une efficacité remarquable en clinique pour prévenir le rejet de greffes (Zenapax®), pour prévenir les infections respiratoires basses dues au virus respiratoire syncytial (VRS) (Synagis®), pour traiter des tumeurs d’origine hématopoïétiques (Mylotarg®, Campath®) ou des tumeurs solides (Herceptine®, Avastin®) et pour traiter des maladies du système immunitaire, qu’il s’agisse d’allergies (Xolair®) ou de maladies auto-immunes telles que le psoriasis (Raptiva®) ou la sclérose en plaque (Tysabri®). L’efficacité d’un anticorps humanisé en clinique dépend bien évidement de l’antigène reconnu par l’anticorps et de l’impact qu’aura la liaison de l’anticorps à l’antigène cible. Lorsque l’anticorps a pour rôle essentiel de transporter sur la cellule cible un agent (radio-élément, agent cytotoxique…) lié à l’anticorps, l’activité clinique de l’anticorps dépend également de l’activité de l’agent transporté par l’anticorps, qui est dit « armé ». Dans le cas du Mylotarg® utilisé pour le traitement des leucémies myéloïdes aigues (LAM) en rechute, l’antigène cible, une lectine (CD33), est présent à la surface de 80 % des cellules de LAM ; l’anticorps est couplé à la calichéamicine qui entraîne une apoptose des cellules malignes par cassure des brins d’ADN. Le mécanisme d’action de ces anticorps armés est donc simple, puisqu’il se réduit à un rôle de transport de l’arme.
Les mécanismes d’action des anticorps humanisés utilisés seuls (anticorps « nus ») sont en revanche moins clairs, comme l’ont écrit Alan Houghton et David Scheinberg en 2000, dans Nature Medicine : « De façon remarquable, après plus de 15 années d’expérience de traitement du cancer avec les anticorps monoclonaux, nous n’avons pas d’idée définitive sur les mécanismes essentiels à leur activité clinique » [13] ; cette phrase reste d’actualité 5 ans plus tard. Classiquement, les mécanismes majeurs sont la cytotoxicité cellulaire dépendante de l’anticorps (CCDA), la cytotoxicité dépendante du complément (CDC), l’initiation de signaux bloquant la croissance ou la survie [13] et l’internalisation de récepteurs de surface cellulaire par l’anticorps. Cette internalisation entraînerait la dégradation du récepteur et inhiberait les voies de transduction des signaux activées par le récepteur. Ce dernier mécanisme a été évoqué pour l’Herceptine®, qui cible le récepteur HER2. Plus récemment, un autre mécanisme a été évoqué pour l’Herceptine® : l’internalisation du récepteur HER2 aboutirait à une augmentation du nombre de peptides dérivés de HER2 et apprêtés pour être présentés par les molécules du complexe majeur d’histocompatibilité (CHM) de classe I : cette augmentation de la présentation stimulerait l’activité cytotoxique de cellules T [14].
Pour les anticorps nus, et quel que soit leur mécanisme d’action, leur efficacité clinique est maintenant largement établie, y compris dans des situations cliniques difficiles comme les cancers du sein métastatiques. Chez les patientes qui ont un cancer du sein métastatique qui surexprime le récepteur HER2, l’addition de l’Herceptine® à la chimiothérapie apporte un réel bénéfice clinique, avec 50 % de réponse objective [15]. Il faut cependant souligner que seules 5 % à 10 % des patientes portant un cancer du sein métastatique surexpriment fortement le récepteur HER2 et peuvent donc être traitées avec l’Herceptine®. Le pourcentage faible de patientes susceptibles de bénéficier de ce traitement et le coût élevé de celui-ci justifie que son utilisation soit conditionnée par les résultats de tests biologiques pour sélectionner les patientes ayant cette surexpression de HER2 [16]. Pour d’autres pathologies comme l’asthme, l’efficacité clinique de l’Omalizumab (Xolair®) a été démontrée chez un pourcentage plus élevé de malades. Cet anticorps humanisé, dirigé contre les IgE, est utilisé dans les cas d’asthmes modérés à sévères en cas d’échec de la corticothérapie classique [17] : il réduit de 38 % l’aggravation de l’asthme [18]. À nouveau, ce traitement coûteux est réservé aux situations cliniques documentées par des tests cutanés ou sanguins.
Des effets adverses parfois imprévisibles
L’exemple de l’anticorps humanisé Tysabri® est une illustration remarquable de l’efficacité clinique d’un anticorps humanisé et des effets adverses parfois imprévisibles d’un tel anticorps. Le Tysabri® (nom générique du natalizumab), est un anticorps dirigé contre la chaîne α4 des intégrines, récepteurs de surface cellulaires composés d’une chaîne α et d’une chaîne β qui jouent un rôle important dans l’adhérence des leucocytes à l’endothélium et dans leur migration dans le parenchyme. En particulier, il avait été montré chez le rat que le natalizumab bloquait l’adhésion des leucocytes à l’endothélium cérébral, prévenait leur accumulation dans le système nerveux central et prévenait le développement de l’encéphalite aiguë expérimentale, modèle animal de la sclérose en plaques (SEP) [19]. La sous-unité α4 de l’intégrine semblait donc être une cible pertinente pour le traitement de la sclérose en plaque avec un anticorps humanisé [20]. Les essais cliniques réalisés chez les sujets présentant une SEP avec des poussées rémittentes ont montré une diminution du nombre de lésions cérébrales et de poussées [20]. Dans une étude menée chez 213 patients, les effets adverses les plus sérieux étaient des réactions anaphylactiques (urticaire, bronchospasme) rapidement réversibles. Il est cependant notable que 11 % des sujets présentaient des anticorps humains anti-anticorps humanisé [21]. Devant l’efficacité de l’anticorps et sa tolérabilité acceptable, son utilisation clinique pour le traitement de formes rémittentes de SEP a été approuvée par la FDA le 23 novembre 2004 ; mais 3 mois plus tard, le 28 février 2005, l’agrément de la FDA était suspendu à la suite du décès de deux patients traités par le Tysabri® et l’interféron-β1a. Un troisième décès a été récemment rapporté chez un patient traité par le Tysabri® seul pour une maladie de Crohn. Ces trois décès sont dus a une leuco-encéphalopathie progressive multifocale, une maladie démyélinisante du système nerveux central associé à l’activation du virus JC, un polyoma virus humain normalement latent chez plus de 80 % des sujets sains [22]. Cet effet adverse fatal met en lumière de façon dramatique les difficultés de la modulation du système immunitaire avec des médicaments actifs tels que les anticorps humanisés. Pour éviter toute confusion, il faut souligner que cet effet adverse est directement lié à la nature de la cible de l’anticorps humanisé, et non à l’humanisation de l’anticorps.
Avenir des anticorps humanisés en clinique
Actuellement, plus de 40 anticorps humanisés sont en essais cliniques [5] et au cours des 6 premiers mois de 2005, 62 articles rapportent les résultats d’essais cliniques avec des anticorps humanisés. Si de nombreux essais ont pour objectif de définir de nouvelles indications pour les anticorps déjà utilisés en clinique, certains sont réalisés avec des anticorps humanisés dirigés contre de nouveaux antigènes : anticorps dirigé contre le récepteur de l’IL6 pour le traitement de la maladie de Crohn, anticorps anti-ACE couplé à l’ytrium 90 pour le traitement des cancers médullaires de la thyroïde avancés, anticorps dirigé contre l’intégrine α4β7 pour le traitement de rectocolite hémorragique sévère, pour ne citer que ces exemples. Les résultats observés avec l’anticorps anti-intégrine α4β7, dénommé MLNO2, sont particulièrement intéressants au moment où des effets adverses fatals ont été observé avec le Tysabri®, anticorps anti-chaîne α4 des intégrines [22]. En premier lieu, le MLNO2 conduit à une rémission clinique chez 32 % à 33 % des patients en fonction de la dose d’anticorps, alors que seuls 12 % des patients sont en rémission dans le groupe placebo, ce qui établit l’activité clinique de cet anticorps [23]. De plus, en dehors d’un cas d’infection à cytomégalovirus qui s’est amélioré sans thérapie antivirale, il n’a pas été observé d’infections virales chez les 118 patients traités, infections virales qui sont désormais l’effet adverse le plus redouté chez les patients traités avec un anticorps anti-intégrine. Cette observation s’expliquerait par la spécificité de l’anticorps MLNO2 pour l’hétérodimère α4β7, intégrine impliquée essentiellement dans le recrutement des leucocytes dans le tractus gastro-intestinal. L’anticorps MLNO2 bloque donc sélectivement l’intégrine α4β7, tandis que le Tysabri® bloque toutes les intégrines composées d’une chaîne α4, et notamment l’intégrine α4β1 qui permet aux leucocytes de se fixer sur l’endothélium de nombreux tissus. Si les résultats observés avec l’anticorps humanisé MLNO2 sont confirmés, ils souligneront l’importance de la spécificité fine d’un anticorps humanisé et rappelleront que cette spécificité conditionne non seulement l’activité thérapeutique d’un anticorps humanisé, mais aussi les effets adverses qu’il peut engendrer.
Les résultats observés avec l’anticorps humanisé MLNO2 soulignent également la fréquence des anticorps humains anti-anticorps humanisés (AHAH) induits par un anticorps humanisé (44 % des malades traités avec l’anticorps MLNO2 présentent des AHAH, par exemple), et leur impact sur l’efficacité de l’anticorps : de fait, chez les patients sans AHAH, ou avec un taux faible d’AHAH, le pourcentage de rémission est de 42 %, contre 12 %, c’est-à-dire un taux proche de celui du groupe placebo (14 %), chez les sujets ayant un titre élevé d’AHAH.
Puisque les anticorps humanisés induisent encore fréquemment une réponse immune anti-anticorps, il semble logique de penser que l’avenir des anticorps humanisés est d’être remplacé par des anticorps totalement humains sensés ne plus être immunogènes. L’exemple de l’Humira® (nom générique de l’adalimubab), premier et seul anticorps humain à avoir reçu l’aval de la FDA pour une utilisation clinique, montre que les anticorps humains demeurent immunogènes. L’Humira® est un anticorps anti-TNFα produit par la technique de sélection guidée [24]. Brièvement, cette technique utilise les séquences variables de l’anticorps murin pour guider la sélection des séquences variables humaines [25], grâce à des librairies d’anticorps chimériques entre la séquence VL ou VH murine et de multiples séquences VL ou VH humaines. Ces séquences chimériques sont présentées à la surface de phages filamenteux (phage display) et la séquence murine, par exemple VH, couplée à de multiples séquences VL humaines, sert à guider la sélection de la séquence VL humaine se liant à l’antigène. La séquence humaine VL sélectionnée est alors liée à de multiples séquences VH humaines et les phages filamenteux qui présentent ces nouveaux anticorps chimériques à l’antigène servent à sélectionner la séquence VH humaine se liant à l’antigène : l’Humira®, produit sur ce principe, est finalement la version entièrement humaine d’un anticorps murin anti-TNFα. Utilisé pour le traitement de la polyarthrite rhumatoïde, cet anticorps induit encore une réponse immune anti-anticorps humain chez 12 % des sujets traités [26] : il n’est donc pas certain que les anticorps humains remplacent immédiatement les anticorps humanisés, qui vont continuer à être de plus en plus utilisés au cours des prochaines années comme continuent à l’être les anticorps chimériques.
Deux raisons principales expliquent le maintien des trois formats d’anticorps (chimériques, humanisés et humains) au cours des prochaines années. La première est l’intérêt clinique de ces anticorps indépendamment de leur format : en oncologie par exemple, c’est un anticorps chimérique, le Rituxan®, qui reste l’anticorps le plus utilisé (traitement des lymphomes non hodgkiniens). Surtout, la question de l’immunogénicité de chaque format d’anticorps reste encore ouverte, et il est sans doute trop simple de penser qu’un anticorps plus humain sera toujours moins immunogène [27] : en effet, les régions CDR de tous ces anticorps, qu’ils soient chimériques, humanisés ou humains, sont uniques et peuvent contenir de façon inhérente des déterminants antigéniques potentiellement immunogéniques [28]. Surtout, l’apparition d’anticorps humains anti-anticorps, si elle dépend du format de l’anticorps, dépend aussi de la quantité injectée, du nombre d’injections, de la voie et du mode d’injection, de la nature de l’antigène cible et de l’immunocompétence du patient [29]. De plus, il a été observé que l’apparition d’anticorps humains anti-anticorps n’est pas toujours délétère et peut parfois avoir des effets bénéfiques [30, 31]. Enfin, il faut reconnaître que, entre laboratoires, il existe des discordances importantes dans la détection et dans la quantification des anticorps humains anti-anticorps [32], et que l’existence de ces discordances complique la comparaison de l’immunogénicité entre les anticorps de différents formats.
Finalement, quel que soit le format des anticorps, une voie d’avenir consiste toujours à diminuer leur immunogénicité. Récemment, des approches in silico ont été développées pour prédire les épitopes T potentiels des anticorps humanisés ou humains. Par analyse informatique des séquences d’immunoglobulines, les épitopes susceptibles de se lier fortement aux molécules de classe II du CMH sont prédits, et les régions correspondantes retirées par mutagenèse dirigée [26]. En théorie, ces modifications devraient diminuer l’immunogénicité, même si ce concept reste à être validé en clinique.
Conclusions
Aujourd’hui, huit anticorps humanisés sont utilisés en clinique chez un grand nombre de patients, ce qui démontre que leur utilisation en thérapeutique est un concept largement validé et, pour reprendre le titre d’un article de Nature, que « les balles magiques ont touché leur cible » [33]. L’activité thérapeutique de ces anticorps s’accompagne parfois d’effets adverses qui ont justifié l’arrêt rapide de l’utilisation clinique d’un anticorps et l’arrêt des essais cliniques d’autres anticorps. Cependant, le choix pertinent de l’antigène cible reconnu par un anticorps pourrait permettre d’éviter certains de ces effets secondaires. Au cours des années à venir, les techniques d’ingénierie des anticorps humanisés devraient encore évoluer pour diminuer leur immunogénicité et réduire le coût de leur production. Ainsi, les anticorps humanisés répondront aux attentes de l’industrie pharmaceutique qui prédit qu’ils deviendront une classe majeure de médicaments, notamment pour le traitement des cancers [34, 35]. Surtout, ils répondront aux besoins des médecins et aux demandes d’un nombre grandissant de malades, concrétisant ainsi le rêve de Paul Ehrlich.
Appendices
Remerciements
Les auteurs remercient Agnès Bellet et Julie Dufour pour leur assistance éditoriale.
Références
- 1. Ehrlich P Herta CA, Shigas K. Über einige Verwendungen der Naphtochinosuflsaure. Z Physiol Chem 1904 ; 61 : 379-92.
- 2. Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975 ; 256 : 495-7.
- 3. Miller RA, Maloney DG, Warnke R, Levy R. Treatment of B-cell lymphoma with monoclonal anti-idiotype antibody. N Engl J Med 1982 ; 306 : 517-22.
- 4. Hwang WY, Foote J. Immunogenicity of engineered antibodies. Methods 2005 ; 36 : 3-10.
- 5. Qu Z, Griffiths GL, Wegener WA, et al. Development of humanized antibodies as cancer therapeutics. Methods 2005 ; 36 : 84-95.
- 6. Morrison SL, Johnson MJ, Herzenberg LA, Oi VT. Chimeric human antibody molecules : mouse antigen-binding domains with human constant region domains. Proc Natl Acad SciUSA 1984 ; 81 : 6851-5.
- 7. Jones PT, Dear PH, Foote J, et al. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature 1986 ; 321 : 522-5.
- 8. Tsurushita N, Hinton PR, Kumar S. Design of humanized antibodies : from anti-Tac to Zenapax. Methods 2005 ; 36 : 69-83.
- 9. Tamura M, Milenic DE, Iwahashi M, et al. Structural correlates of an anticarcinoma antibody : identification of specificity-determining residues (SDRs) and development of a minimally immunogenic antibody variant by retention of SDRs only. J Immunol 2000 ; 164 : 1432-41.
- 10. Zhang W, Feng J, Li Y, et al. Humanization of an anti-human TNF-alpha antibody by variable region resurfacing with the aid of molecular modeling. Mol Immunol 2005 ; 42 : 1445-51.
- 11. Roguska MA, Pedersen JT, Keddy CA, et al. Humanization of murine monoclonal antibodies through variable domain resurfacing. Proc Natl Acad Sci USA 1994 ; 91 : 969-73.
- 12. Padlan EA. A possible procedure for reducing the immunogenicity of antibody variable domains while preserving their ligand-binding properties. Mol Immunol 1991 ; 28 : 489-98.
- 13. Houghton AN, Scheinberg DA. Monoclonal antibody therapies-a ‘constant’ threat to cancer. Nat Med 2000 ; 6 : 373-4.
- 14. Zum Buschenfelde CM, Hermann C, Schmidt B, et al. Antihuman epidermal growth factor receptor 2 (HER2) monoclonal antibody trastuzumab enhances cytolytic activity of class I-restricted HER2-specific T lymphocytes against HER2-overexpressing tumor cells. Cancer Res 2002 ; 62 : 2244-7.
- 15. Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001 ; 344 : 783-92.
- 16. Elkin EB, Weinstein MC, Winer EP, et al. HER-2 testing and trastuzumab therapy for metastatic breast cancer : a cost-effectiveness analysis. J Clin Oncol 2004 ; 22 : 854-63.
- 17. Ames SA, Gleeson CD, Kirkpatrick P. Omalizumab. Nat Rev Drug Discov 2004 ; 3 : 199-200.
- 18. Holgate ST, Djukanovic R, Casale T, Bousquet J. Anti-immunoglobulin E treatment with omalizumab in allergic diseases : an update on anti-inflammatory activity and clinical efficacy. Clin Exp Allergy 2005 ; 35 : 408-16.
- 19. Yednock TA, Cannon C, Fritz LC, et al. Prevention of experimental autoimmune encephalomyelitis by antibodies against alpha4 beta1 integrin. Nature 1992 ; 356 : 63-6.
- 20. Noseworthy JH, Kirkpatrick P. Natalizumab. Nat Rev Drug Discov 2005 ; 4 : 101-2.
- 21. Miller DH, Khan OA, Sheremata WA, et al. A controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med 2003 ; 348 : 15-23.
- 22. Van Assche G, Van Ranst M, Sciot R, et al. Progressive Multifocal leukoencephalopathy after natalizumab therapy for Crohn’s disease. N Engl J Med online 9 juin 2005.
- 23. Feagan BG, Greenberg GR, Wild G, et al. Treatment of ulcerative colitis with a humanized antibody to the alpha4 beta7 integrin. N Engl J Med 2005 ; 352 : 2499-507.
- 24. Osbourn J, Groves M, Vaughan T. From rodent reagents to human therapeutics using antibody guided selection. Methods 2005 ; 36 : 61-8.
- 25. Jespers LS, Roberts A, Mahler SM, et al. Guiding the selection of human antibodies from phage display repertoires to a single epitope of an antigen. Biotechnology (NY) 1994 ; 12 : 899-903.
- 26. Wu H, Dall’Acqua WF. Humanized antibodies and their applications. Methods 2005 ; 36 : 1-2.
- 27. Anderson PJ. Tumor necrosis factor inhibitors : clinical implications of their different immunogenicity profiles. Semin Arthritis Rheum 2005 ; 34 : 19-22.
- 28. Ritter G, Cohen LS, Williams C Jr, et al. Serological analysis of human anti-human antibody responses in colon cancer patients treated with repeated doses of humanized monoclonal antibody A33. Cancer Res 2001 ; 61 : 6851-9.
- 29. Kuus-Reichel K, Grauer LS, Karavodin LM, et al. Will immunogenicity limit the use, efficacy, and future development of therapeutic monoclonal antibodies ? Clin Diagn Lab Immunol 1994 ; 1 : 365-72.
- 30. DeNardo GL, Bradt BM, Mirick GR, DeNardo SJ. Human antiglobulin response to foreign antibodies : therapeutic benefit ? Cancer Immunol Immunother 2003 ; 52 : 309-16.
- 31. Mirick GR, Bradt BM, Denardo SJ, Denardo GL. A review of human anti-globulin antibody (HAGA, HAMA, HACA, HAHA) responses to monoclonal antibodies. Not four letter words. Q J Nucl Med Mol Imaging 2004 ; 48 : 251-7.
- 32. Kimball JA, Norman DJ, Shield CF, et al. The OKT3 antibody response study : a multicentre study of human anti-mouse antibody (HAMA) production following OKT3 use in solid organ transplantation. Transpl Immunol 1995 ; 3 : 212-21.
- 33. Gura T. Therapeutic antibodies : magic bullets hit the target. Nature 2002 ; 417 : 584-6.
- 34. Reichert JM. Monoclonal antibodies in the clinic. Nat Biotechnol 2001 ; 19 : 819-22.
- 35. Featherstone J, Griffiths S. From the analyst’s couch. Drugs that target angiogenesis. Nat Rev Drug Discov 2002 ; 1 : 413-4.
List of figures
Figure 1
Pourcentage d'anticorps induisant une réponse immune anti-anticorps.
Une réponse est qualifiée de négligeable lorsqu'elle se produit chez moins de 2 % des patients, tolérable lorsqu'elle se produit chez 2 % à 15% des patients et marquée lorsqu'elle se produit chez plus de 15% d'entre eux. AHAM : anticorps humains anti-anticorps murins ; AHAC : anticorps humains anti-anticorps chimériques ; AHAH : anticorps humains anti-anticorps humanisés (d'après [4]).
Figure 2
Formes d’anticorps monoclonaux utilisés en thérapeutique.
Anticorps chimérique : les régions constantes des chaînes lourdes et légères des immunoglobulines murines sont remplacées par des régions constantes humaines. Anticorps humanisé : les régions hypervariables (complementary determining region, CDR) d’un anticorps humain sont remplacées par des régions CDR d’origine murine, qui constituent le site de liaison de l’antigène. Cependant, certains acides aminés situés dans les régions adjacentes aux régions CDR (régions charpentes, ou FR pour framework) jouent un rôle majeur dans la structure du site de liaison de l’antigène. Pour conserver cette structure au cours de l’humanisation, l’anticorps accepteur reçoit non seulement les régions CDR de l’anticorps murin donneur, mais aussi les acides aminés de la région charpente de l’anticorps donneur.
Figure 3
Protocoles d’immunisation de la région variable d’une chaîne légère.
CDR : région déterminante complémentaire ; SDR : résidu déterminant spécifique (acide aminé des régions CDR critique dans l’interaction antigène-anticorps) : CDR abrégés : portions des CDR contenant les résidus SDR (d’après [9]).
List of tables
Tableau I
Anticorps monoclonaux humanisés utilisés en clinique.
VRS : virus respiratoire syncitial ; HER2 : récepteur Erb2 du facteur de croissance épidermique humain ; VEGF : vascular endothelium growth factor ; IL-2 : interleukine-2 ; CCDA : cytotoxicité cellulaire dépendante de l’anticorps ; CDC : cytotoxicité dépendante du complément ; LNH : lymphome non hodgkinien ; LAM : leucémie aiguë myéloïde ; LLC : leucémie lymphoïde chronique. *Date d’approbation par les autorités américaines (Food and drug administration, FDA).