Article body
La lèpre, ou maladie de Hansen, est une maladie infectieuse chronique, provoquée par le bacille Mycobacterium leprae, qui atteint principalement les parties les plus froides du corps, en particulier la peau, les segments superficiels des nerfs périphériques, les voies respiratoires supérieures, les testicules et les yeux [1]. La lèpre est, parmi les maladies transmissibles, celle qui cause le plus d’incapacités physiques permanentes, et les difformités terribles dues à la forme avancée de la maladie ont largement contribué aux préjugés profonds à l’origine du rejet des malades et de leurs proches par la société. Heureusement, les avancées thérapeutiques décisives des années 1980 ont conduit, en 1991, l’Organisation Mondiale de la Santé à faire de l’élimination de la lèpre un de ses objectifs majeurs pour la fin de l’année 2000. De fait, parmi les 122 pays considérés comme endémiques en 1985, 108 ont effectivement éradiqué la maladie, et c’est au total plus de 12 millions de patients qui ont été guéris grâce à la polychimiothérapie [2]. Toutefois, parodiant Mark Twain, on peut se demander si « les rumeurs sur la disparition de la lèpre n’ont pas été grandement exagérées ». En effet, la polychimiothérapie n’a pas eu d’impact sur le cycle de transmission de la maladie et tandis que l’on diagnostiquait environ 600 000 nouveaux cas de lèpre en 1991, ce sont 760 000 nouveaux cas qui ont été dépistés dans le monde en 2001. La cause de la stagnation de cette incidence à un niveau élevé, alors que le réservoir naturel de M. leprae est limité à l’homme et le traitement efficace, n’est pas connue, indiquant que des aspects importants de la relation complexe entre l’agent pathogène et son hôte humain restent à découvrir.
Après un contact prolongé avec M. leprae, une grande majorité des individus développe une immunité efficace sans expression clinique tandis que moins de 5 % vont présenter un large spectre de manifestations cliniques [1]. Schéma-tiquement, environ 60 % des nouveaux cas sont des formes dites paucibacillaires, également appelées tuberculoïdes, caractérisées par un petit nombre de lésions cutanées, l’absence de bacilles histologiquement détectables dans ces lésions et une réponse immune cellulaire spécifique adaptée. Les 40 % de patients restants expriment des formes multibacillaires, ou lépromateuses, caractérisées par de nombreuses lésions cutanées gorgées de M. leprae et une altération de l’immunité cellulaire spécifique. Cette pathogénie de la lèpre en deux étapes est apparemment le reflet d’un contrôle génétique à deux niveaux : certains gènes influencent la susceptibilité intrinsèque à la lèpre, on parle de lèpre per se, c’est-à-dire indépendante de la forme clinique, et d’autres influencent une forme clinique spécifique. Tandis que certains gènes avaient été impliqués soit dans le contrôle des formes cliniques - en particulier des gènes de la région HLA, le gène TLR2 (toll like receptor) et un locus situé sur le chromosome 10 (10p13) (pour revues, voir [3, 4]) - soit dans celui de la phase précoce de l’infection - le gène NRAMP1 [5] -, la contribution génétique à la lèpre per se restait à élucider. Dans ce but, nous avions mis en place depuis plusieurs années une grande étude d’épidémiologie génétique dans la région d’Ho Chi Minh Ville, au Vietnam.
Dans un premier échantillon de 86 familles incluant au moins deux enfants atteints de lèpre, et des proportions similaires de cas pauci- et multibacillaires, nous avons réalisé une analyse de liaison génétique par criblage complet du génome [6]. Son principe général consiste à rechercher des régions chromosomiques qui ségrègent de façon non aléatoire avec le phénotype d’intérêt - la lèpre per se - au sein des familles afin d’identifier dans ces régions un ou quelques gènes d’intérêt. Cette première étude a montré une liaison très significative entre la lèpre per se et une région chromosomique située en 6q25 (lod score = 4,31, p < 5.10‑6), résultat que nous avons confirmé dans un échantillon indépendant de 208 familles. Dans le contexte des maladies complexes, une limite de l’analyse de liaison par criblage du génome est le pouvoir de résolution. En effet, il est fréquent que la région chromosomique identifiée s’étende sur plusieurs dizaines de centimorgans. Ainsi, dans notre étude, l’intervalle de confiance à 90 % pour le positionnement du locus de susceptibilité à l’origine du signal de liaison était compris entre deux marqueurs distants de 6,4 mégabases (Mb, Figure 1A) et contenait 31 gènes ayant des fonctions connues ou prédites (Figure 1B). Le second objectif consistait donc à réduire cet intervalle par une approche dite de « cartographie par étude du déséquilibre de liaison » ou LD mapping. Tandis que l’analyse de liaison s’intéresse à la proportion d’allèles marqueurs partagée par des enfants atteints au sein d’une même fratrie, le criblage par déséquilibre de liaison repose sur l’observation que d’une famille à l’autre le même allèle est partagé par des enfants atteints. Explicitement, déséquilibre de liaison entre un marqueur et le locus causal signifie liaison génétique et association allélique, c’est-à-dire qu’un allèle donné du marqueur est préférentiellement associé à l’allèle de susceptibilité du locus causal, ce qui est en particulier le cas lorsque l’allèle de susceptibilité fait partie des allèles marqueurs testés [4].
Figure 1
Étude d’association de la susceptibilité à la lèpre per se dans la région chromosomique 6q25.
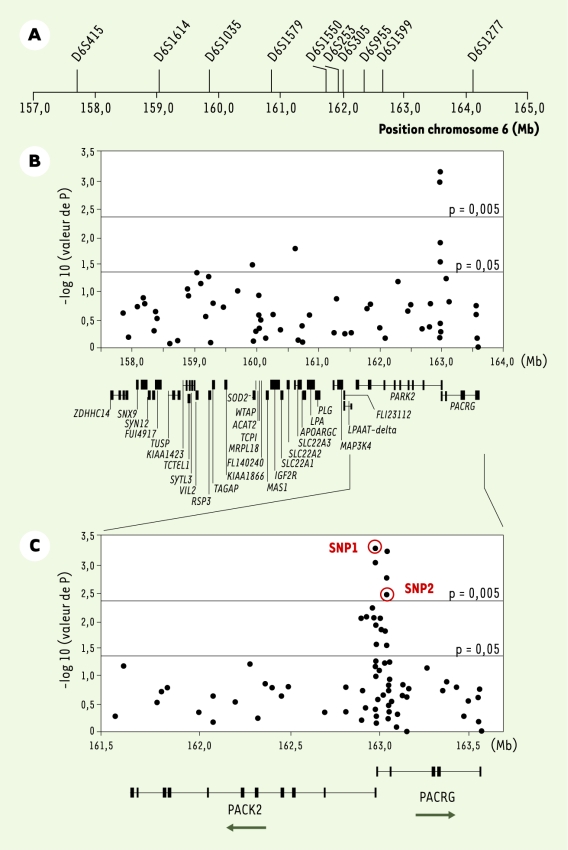
A. Marqueurs microsatellites utilisés pour l’analyse de liaison génétique fine. Les flèches indiquent la position des marqueurs en mégabases (Mb) sur le chromosome 6 (http://ncbi.nlm.nih.gov). Le pic de lod score à 4,31 est observé entre D6S1579 et D6S1550. L’intervalle de confiance à 90 % pour le positionnement du locus de susceptibilité entraînant ce signal de liaison couvre une région de 6,4 Mb entre les marqueurs D6S415 et D6S1277. B. Étude d’association familiale de la lèpre avec 64 SNP (single nucleotide polymorphisms) couvrant l’intervalle de confiance à 90 %. Le degré de signification (p) de l’association de chacun des SNP avec la lèpre per se est exprimé sur l’axe des ordonnées [en -log10(p)] en fonction de la localisation des SNP (axe des abscisses en Mb). La localisation et la structure génomique des 31 gènes connus ou prédits de la région sont également données. Les exons sont représentés par des petites barres verticales et les introns par de fines lignes horizontales. Pour les gènes avec de petits introns, les exons sont fusionnés en un rectangle noir. C. Expansion de la région chromosomique couvrant les gènes PARK2 et PACRG. Le degré de signification de chacun des 81 SNP testés est donné en ordonnée en fonction de leur position chromosomique en Mb. La structure génomique et le sens de la transcription sont représentés pour les deux gènes. Les cercles rouges identifient les deux SNP qui suffisent à capturer toute l’information d’association entre cette région et la lèpre per se.
Dans un échantillon de 197 familles, nous avons saturé les 6,4 mégabases du pic de liaison par un panel de 64 marqueurs dialléliques, communément appelés SNP (single nucleotide polymorphisms), et nous avons effectué une analyse de cartographie par étude du déséquilibre de liaison (LD mapping) (Figure 1B) [7]. Parmi les six SNP significativement associés à la lèpre per se, quatre, dont les deux plus significatifs, étaient situés dans une région chevauchant le promoteur commun au gène PARK2, dont des mutations sont responsables de certaines formes juvéniles de maladie de Parkinson, et au gène coréglé PACRG. Par séquençage comparatif et criblage des bases de données, un ensemble de 81 SNP saturant cette région d’environ 2 Mb (Figure 1C) a été développé. Dix-neuf SNP furent trouvés associés à la lèpre, tous localisés dans un bloc s’étendant sur près de 80 kilobases et incluant la région régulatrice commune aux deux gènes PARK2 et PACRG. Afin d’identifier quels SNP étaient à l’origine du signal d’association, nous avons réalisé une « régression logistique conditionnelle multivariée » [8] qui a montré que deux SNP PARK2-e01(‑2599C/T) et rs1040079(C/T), nommés SNP1 et SNP2, suffisaient à capturer toute l’information d’association entre la région associée et la lèpre (cercles rouges sur la Figure 1C). En raison de l’important déséquilibre de liaison entre ces deux SNP, seuls trois haplotypes (sur quatre possibles) furent observés. Ces trois haplotypes définissaient six génotypes (Figure 2). Les sujets homozygotes pour l’haplotype C-T (contenant l’allèle C du SNP1 et l’allèle T du SNP2) présentaient le risque le plus faible de développer la lèpre et définirent la population de référence. Par comparaison avec ces individus, les porteurs d’au moins une copie de l’haplotype à haut risque T-C avaient un risque environ cinq fois supérieur de développer la maladie tandis que les porteurs d’au moins une copie de l’haplotype à risque intermédiaire (T-T), sans haplotype à haut risque, avait un risque multiplié par trois. L’association fut reproduite dans un échantillon de 587 cas et 388 témoins non apparentés originaires de Rio de Janeiro au Brésil (p < 5.10-7) identifiant ainsi ces variants dans la région régulatrice partagée par PARK2 et PACRG comme des facteurs de risque universels pour la lèpre per se. Enfin, nous avons montré que ces deux gènes sont exprimés dans les macrophages et les cellules de Schwann qui sont les principales cellules cibles de M. leprae, ce qui conforte les résultats de l’épidémiologie génétique.
Figure 2
Association entre la lèpre per se et les génotypes définis à partir des trois haplotypes PARK2-e01(-2599)-rs1040079.
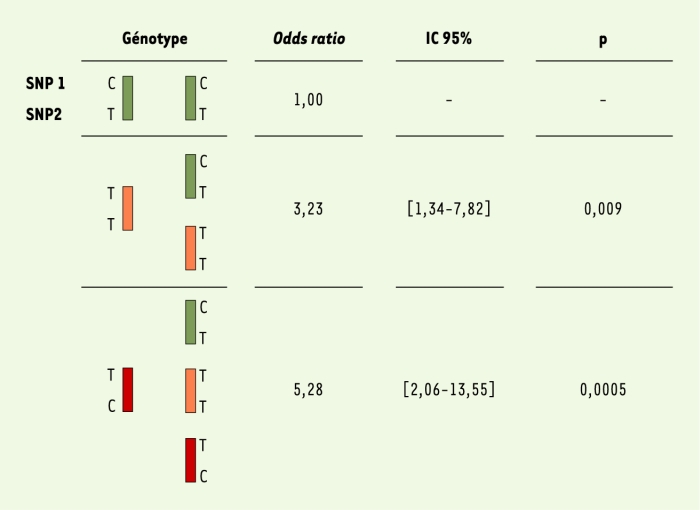
PARK2-e01(-2599) et rs1040079 sont respectivement et arbitrairement nommés SNP1 et SNP2. Les odds ratios avec leurs intervalles de confiance à 95 % (IC 95 %) ont été estimés par régression logistique conditionnelle [8].
Fonctionnellement, les deux gènes sont impliqués dans l’une des cascades majeures de dégradation des protéines cellulaires, la cascade d’ubiquitinylation-protéolyse. Grâce à son activité E3 ubiquitine-ligase, PARK2 reconnaît des substrats spécifiques et induit leur poly-ubiquitinylation qui est un signal de ciblage vers la voie de dégradation par le protéasome [9]. Ainsi, des mutations dans PARK2 sont responsables de la forme juvénile de maladie de Parkinson, probablement par accumulation de substrats de PARK2 conduisant à la dégénérescence des neurones dopaminergiques de la voie nigro-striatale. Cette cascade ubiquitine-protéasome n’avait jamais été évoquée dans ce contexte de l’infection par M. leprae, et elle ouvre des perspectives totalement nouvelles pour la physiopathologie de la maladie. Actuellement, des études fonctionnelles sont en cours afin de caractériser la nature exacte de l’interaction entre ces mécanismes particuliers de dégradation protéique et l’infection par M. leprae. Dans un avenir proche, l’identification des sujets prédisposés devrait jouer un rôle important dans le développement et l’évaluation des programmes de contrôle de la maladie et dans l’élaboration de nouvelles stratégies préventives et thérapeutiques. Ainsi, des approches préventives qui s’avèrent peu efficaces ou techniquement irréalisables à l’échelle d’une population dans son ensemble, pourraient présenter un intérêt majeur lorsqu’elles sont ciblées sur un groupe d’individus à haut risque de développer la maladie. À plus long terme, ces travaux devraient ouvrir de nouvelles possibilités thérapeutiques orientées vers la restauration d’une réponse adaptée de l’hôte lors de la rencontre avec l’agent pathogène. Enfin, cette découverte soulève l’hypothèse fascinante que des anomalies dans la façon dont les cellules gèrent les protéines superflues (comme les protéines devenues inutiles ou reconnues comme étrangères) pourraient être à l’origine de maladies assez communes et très différentes dans leur présentation comme des maladies infectieuses ou des maladies neurodégénératives.
Appendices
Références
- 1. Jacobson RR, Krahenbuhl JL. Leprosy. Lancet 1999 ; 353 :655-60.
- 2. WHO. Leprosy : global situation. Wkly Epidemiol Rec 2000 ;75 :225-232.
- 3. Casanova JL, Abel L. Genetic dissection of immunity to mycobacteria : the human model. Annu Rev Immunol 2002 ;20 : 581-620.
- 4. Alcais A, Abel L. Application of genetic epidemiology to dissecting host susceptibility/resistance to infection illustrated with the study of common mycobacterial infections. In : Bellamy R, ed. Susceptibility to infectious diseases. Cambridge : Cambridge University Press, 2004 :7-45.
- 5. Alcais A, Sanchez FO, Thuc NV, et al. Granulomatous reaction to intradermal injection of lepromin (Mitsuda reaction) is linked to the human NRAMP1 gene in Vietnamese leprosy sibships. J Infect Dis 2000 ; 181 :302-8.
- 6. Mira MT, Alcais A, Van Thuc N, et al. Chromosome 6q25 is linked to susceptibility to leprosy in a Vietnamese population. Nat Genet 2003 ;33 :412-5.
- 7. Mira MT, Alcais A, Nguyen VT, et al. Susceptibility to leprosy is associated with PARK2 and PACRG. Nature 2004 ;427 :636-40.
- 8. Schaid DJ. General score tests for associations of genetic markers with disease using cases and their parents. Genet Epidemiol 1996 ;13 :423-49.
- 9. Corti O, Brice A. La maladie de Parkinson : que nous apprennent les gènes des formes familiales ? Med Sci (Paris) 2003 ;19 :613-9.
List of figures
Figure 1
Étude d’association de la susceptibilité à la lèpre per se dans la région chromosomique 6q25.
A. Marqueurs microsatellites utilisés pour l’analyse de liaison génétique fine. Les flèches indiquent la position des marqueurs en mégabases (Mb) sur le chromosome 6 (http://ncbi.nlm.nih.gov). Le pic de lod score à 4,31 est observé entre D6S1579 et D6S1550. L’intervalle de confiance à 90 % pour le positionnement du locus de susceptibilité entraînant ce signal de liaison couvre une région de 6,4 Mb entre les marqueurs D6S415 et D6S1277. B. Étude d’association familiale de la lèpre avec 64 SNP (single nucleotide polymorphisms) couvrant l’intervalle de confiance à 90 %. Le degré de signification (p) de l’association de chacun des SNP avec la lèpre per se est exprimé sur l’axe des ordonnées [en -log10(p)] en fonction de la localisation des SNP (axe des abscisses en Mb). La localisation et la structure génomique des 31 gènes connus ou prédits de la région sont également données. Les exons sont représentés par des petites barres verticales et les introns par de fines lignes horizontales. Pour les gènes avec de petits introns, les exons sont fusionnés en un rectangle noir. C. Expansion de la région chromosomique couvrant les gènes PARK2 et PACRG. Le degré de signification de chacun des 81 SNP testés est donné en ordonnée en fonction de leur position chromosomique en Mb. La structure génomique et le sens de la transcription sont représentés pour les deux gènes. Les cercles rouges identifient les deux SNP qui suffisent à capturer toute l’information d’association entre cette région et la lèpre per se.
Figure 2
Association entre la lèpre per se et les génotypes définis à partir des trois haplotypes PARK2-e01(-2599)-rs1040079.
PARK2-e01(-2599) et rs1040079 sont respectivement et arbitrairement nommés SNP1 et SNP2. Les odds ratios avec leurs intervalles de confiance à 95 % (IC 95 %) ont été estimés par régression logistique conditionnelle [8].