Abstracts
Résumé
L’analyse à grande échelle des protéines, également appelée analyse protéomique, a parfois été présentée comme un eldorado de la biologie moderne. La quête de l’Eldorado a conduit nombre de conquistadors à s’attaquer à une jungle impénétrable qui fut leur tombeau. Il serait regrettable que les biologistes subissent le même sort, et cet article va s’employer à éclaircir la jungle de l’analyse protéomique. Les grands concepts de l’analyse protéomique seront présentés, ainsi que les technologies actuelles utilisées dans le domaine. Les performances comme les limitations de ces technologies seront exposées, afin de donner au lecteur une vue aussi raisonnée que possible de l’état de l’art dans ce domaine de recherche. Enfin, les grandes applications actuelles et les développements prévisibles de l’analyse protéomique en thérapeutique seront discutés.
Summary
The present paper aims at clarifying some important aspects of proteomics, i.e. the large scale analysis of proteins. To this purpose, the main types of proteomic analyses are presented, i.e. those aiming at determining expression levels and those aiming at unravelling protein-protein interactions networks. Their performances and limitations are outlined, as well as their potential applications in biomedicine, to give an reasoned view of the current state of the art.
Article body
Alors que l’essentiel de la recherche en biologie avait suivi le chemin réductionniste et hypothético-déductif, les dernières années ont été marquées par l’émergence du concept de biologie à grande échelle. Ce concept repose sur le postulat selon lequel les systèmes biologiques fonctionnent comme des réseaux complexes, et qu’une étude à l’échelle la plus grande possible peut apporter un surcroît de connaissances. La conceptualisation de cette approche, qui remonte au début des années 80 [1], a conduit à l’application de cette démarche aux macromolécules biologiques. Du fait de la maturité des outils utilisés pour le clonage et le séquençage de l’ADN, cette molécule a été étudiée la première à l’échelle de génomes entiers, et les progrès ont été impressionnants depuis la première publication de la séquence complète d’un génome [2]. En revanche, si le génome fournit le répertoire de chaque cellule d’un organisme, la réalité cellulaire va dépendre de l’emploi de ce répertoire en termes de choix des gènes exprimés, de niveaux d’expression des produits géniques, et de modulation de la fonctionnalité des protéines, par exemple par des modifications post-traductionnelles. De plus, il apparaît de plus en plus clairement que les protéines ne fonctionnent pas dans la cellule de façon isolée, mais sous forme de complexes multimoléculaires. Ainsi, il devient évident que l’étude des protéines sera plus proche de la physiologie cellulaire. Une branche nouvelle de la biochimie visant à l’étude à grande échelle des protéines s’est donc développée récemment. De même que l’ensemble des gènes d’un organisme forme son génome, l’ensemble des protéines d’une cellule ou d’un tissu forme son protéome, et cette discipline a reçu le nom d’analyse protéomique. Cette discipline se subdivise en deux tendances selon la propriété des protéines qui est examinée en priorité, et l’on distingue ainsi la protéomique d’expression et la protéomique d’interactions.
La protéomique d’expression
Dans cette branche de l’analyse protéomique, on cherche à identifier les gènes exprimés dans une condition donnée, mais aussi à déterminer les quantités de chaque protéine présentes dans l’objet biologique étudié, en prenant autant que possible en compte les différents variants de protéines provenant par exemple de modifications post-traductionnelles. En corollaire, la détermination de la nature et de la localisation des modifications post-traductionnelles est une part importante de cette discipline.
Électrophorèse bidimensionnelle et spectrométrie de masse
Dans sa version classique, la protéomique d’expression couple l’électrophorèse bidimensionnelle à la spectrométrie de masse (Figure 1). Dans ce schéma, l’électrophorèse bidimensionnelle permet de séparer plusieurs centaines de formes protéiques, et en particulier un grand nombre de variants post-traductionnels, avec une dimension quantitative et une capacité unique de séparer des protéines entières. La spectrométrie de masse apporte quant à elle sa capacité à caractériser les protéines ainsi séparées avec force détails, comme l’identification de modifications post-traductionnelles atypiques, et bien sûr la détermination de leur site (pour revue, voir [3]). Si cette technique a fait ses preuves, elle a aussi montré ses limites en termes de spectre d’analyse, par exemple des protéines minoritaires et membranaires [4-6], ou en termes de débit d’analyse et de miniaturisation. Ces limites provenant essentiellement de l’électrophorèse bidimensionnelle, d’autres schémas d’analyse protéomique ont été développés.
Figure 1
Analyse d’un mélange de protéines par électrophorèse bidimensionnelle et spectrométrie de masse.
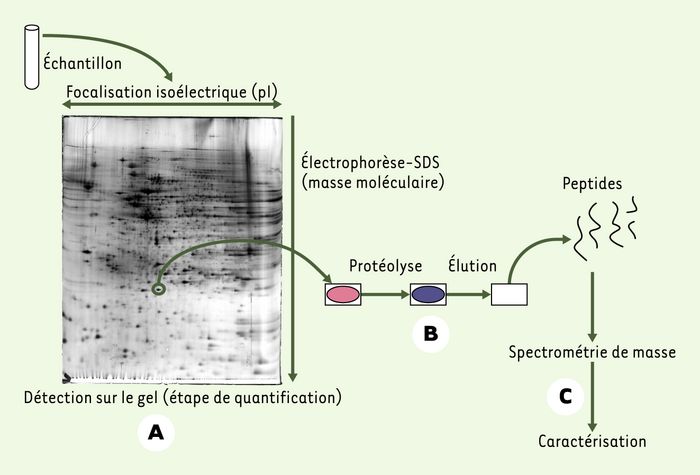
A. Le mélange de protéines est séparé par électrophorèse bidimensionnelle. B. Les taches d’intérêt, observées après coloration, sont digérées pour donner des peptides. C. Ces peptides sont extraits puis analysés par spectromètre de masse. Cette analyse fournit le nom des protéines présentes dans le fragment de gel de départ, ainsi que certaines des modifications post-traductionnelles portées par cette forme protéique.
Chromatographie et spectrométrie de masse
Parmi eux, les schémas fondés sur la séparation chromatographique des protéines méritent d’être cités. Pour des raisons de miniaturisation, ces techniques ne sont pas implantées sous forme de colonne chromatographique, mais sous forme de surfaces d’adsorption planes ou de billes.
Dans la technologie SELDI (surface enhanced laser desorption-ionization) [7], ce qui est retenu sur la surface chromatograpique est analysé en spectrométrie de masse. Le principe consiste donc à déposer un échantillon complexe sur la surface chromatographique, puis à éluer partiellement cet échantillon jusqu’à ce que soit adsorbé un nombre de protéines compatibles avec la résolution du spectromètre de masse. Ce système est utilisé en analyse comparative pour analyser des marqueurs présents dans un échantillon par rapport à un autre. En variant le type de support chromatographique et le schéma d’élution partielle, il est souvent possible de mettre en évidence des marqueurs différentiels. Du fait de sa miniaturisation et de sa simplicité, cette technique est bien adaptée à l’analyse d’échantillons de petite taille comme des biopsies cliniques [8]. Elle pâtit cependant de la difficulté à réaliser une analyse poussée des marqueurs ainsi mis en évidence, et est plutôt adaptée à l’analyse des protéines de petite taille. De plus, du fait même du principe de l’analyse, les pertes en termes de nombre de marqueurs présomptifs sont considérables. Dans un autre schéma, ce sont les éluats successifs de l’échantillon complexe adsorbé sur la bille chromatographique qui sont analysés, ce qui permet une meilleure prise en compte de la complexité de l’échantillon. De plus, l’utilisation de techniques de digestion et d’analyse des peptides est facilitée, ce qui permet une caractérisation détaillée des marqueurs d’intérêt. Ces caractéristiques positives sont contrebalancées par une analyse plus laborieuse, et par le fait que nombre de protéines ne sont pas éluables des supports chromatographiques par un système compatible avec les étapes ultérieures fondées sur la spectrométrie de masse.
Du fait des problèmes rencontrés dans les analyses reposant sur la séparation de protéines entières, ce sont directement les peptides issus du clivage en masse des protéines cellulaires qui sont analysés dans le schéma appelé MUDPIT (multidimensional protein identification technology) (Figure 2). Comme il est rare qu’une protéine, quelle qu’elle soit, ne donne pas au moins un peptide apparaissant dans le spectromètre de masse, cette technique est très performante en termes de spectre des protéines analysées [9], ainsi qu’en termes de détermination de modifications post-traductionnelles [10]. En revanche, elle ne permet pas d’accéder aux quantités des protéines, et encore moins aux quantités relatives des différents variants de protéines. Afin de pallier ce grave défaut, tout en gardant les capacités des techniques fondées sur l’analyse directe des peptides, la technique dite ICAT (isotope coded affinity tag) a été proposée [11] (Figure 3) et validée [12].
Figure 2
Analyse d’un mélange de protéines par la méthode MUDPIT (multidimensional protein identification technology).

A. Le mélange de protéines est directement digéré par la trypsine. B. Le mélange complexe de peptides résultant de cette digestion est ensuite acidifié avec de l’acide acétique (AcOH). C. Le mélange est déposé sur un système composé de deux colonnes chromatographiques en tandem, relié à un spectromètre de masse (MS) en tandem : la première colonne est un échangeur fort de cations (CE), la deuxième une phase inverse (PI). D. L’élution de la colonne échangeuse d’ions par une concentration donnée de sels (NH4HCO3) transfère une partie des peptides vers la colonne de phase inverse. E. Après lavage pour éliminer le sel, la colonne de phase inverse est développée par un gradient d’éthanol (EtOH, 1 heure), et les peptides sortant de la colonne sont analysés par spectrométrie de masse en tandem, ce qui permet de déterminer leur nature, et donc la protéine dont ils proviennent. Une fois l’élution terminée, une étape de salinité plus importante est appliquée pour transférer d’autres peptides vers la colonne de phase inverse ; une vingtaine de ces cycles sont réalisés.
Figure 3
Analyse comparative par la méthode ICAT (isotope coded affinity tag).
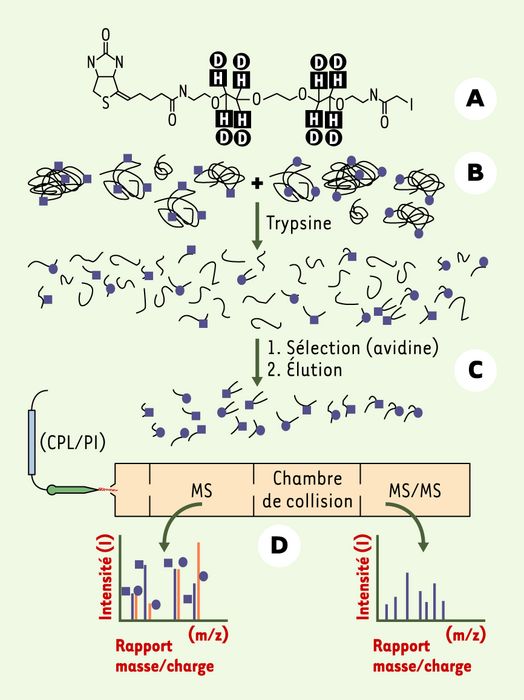
A. Schéma d’une sonde ICAT spécifique des cystéines. Cette sonde est constituée de trois parties : une partie réagissant avec les cystéines (iodoacétamide), une biotine (à gauche) qui permet la sélection par chromatographie d’affinité et un bras espaceur qui peut contenir des hydrogènes (carrés) ou des deutérium (ronds). B. Les deux extraits à comparer sont ensuite marqués, l’un avec la sonde légère (hydrogène), l’autre avec la sonde lourde (deutérium). C. Les extraits sont mélangés et digérés par la trypsine, puis les peptides marqués par la sonde sont sélectionnés par chromatographie d’affinité. D. Les peptides sélectionnés sont analysés par chromatographie liquide (phase inverse) (CPL/PI) couplée en ligne avec un spectromètre de masse (MS) en tandem. Le rapport des signaux correspondant aux peptides identiques marqués par la sonde légère et la sonde lourde, observé dans le premier analyseur du spectromètre de masse, donne le rapport quantitatif des protéines d’origine. Les données de fragmentation des peptides donnent la nature de la protéine d’origine. On notera que les protéines dénuées de cystéines échappent à l’analyse.
Cependant, il faut souligner que ces techniques fondées sur l’analyse des peptides de digestion n’analysent que quelques peptides par protéine. Malgré ses performances impressionnantes, la technique MUDPIT est ainsi incapable de détecter une proportion importante (jusqu’à 50 %) des protéines visualisées par l’approche classique électrophorèse bidimensionnelle-spectrométrie de masse [13]. À l’heure actuelle, aucune technique ni combinaison de techniques ne peut donc prétendre à l’exhaustivité, en particulier en ce qui concerne la détermination des modifications post-traductionnelles. Une analyse par électrophorèse bidimensionnelle concerne au mieux 2 000 formes protéiques, et une analyse MUDPIT 20 000 peptides, chiffres à mettre en regard des 20 000 formes protéiques et des 200 000 peptides tryptiques prédits pour une cellule humaine.
La protéomique d’interactions
Dans cette branche de l’analyse protéomique, l’enjeu est de déterminer les interactions physiques entre les protéines d’une même cellule, et les éventuelles variations de la composition de ces complexes multiprotéiques dans différentes situations biologiques. Pour cela, deux grandes techniques sont utilisées.
L’approche double-hybride
La première approche est celle du double-hybride [14] (Figure 4). Cette technique est maintenant bien établie, et ses défauts, en particulier en termes de spécificité des interactions mises en évidence, sont bien connus. Des progrès récents ont cependant été enregistrés, en utilisant notamment des séquences correspondant à des domaines de protéines, et non à des protéines entières. Le gain de spécificité permet d’envisager des déterminations d’interactions protéine-protéine à l’échelle de cellules entières [15]. Cependant, ce schéma ne permet de déterminer que des interactions binaires entre protéines. De plus, l’influence des modifications post-traductionnelles est sous-évaluée, dans la mesure où seules celles présentes chez l’hôte servant à réaliser les expériences de double-hybride (généralement la levure) peuvent être prises en compte.
Figure 4
L’approche double-hybride.
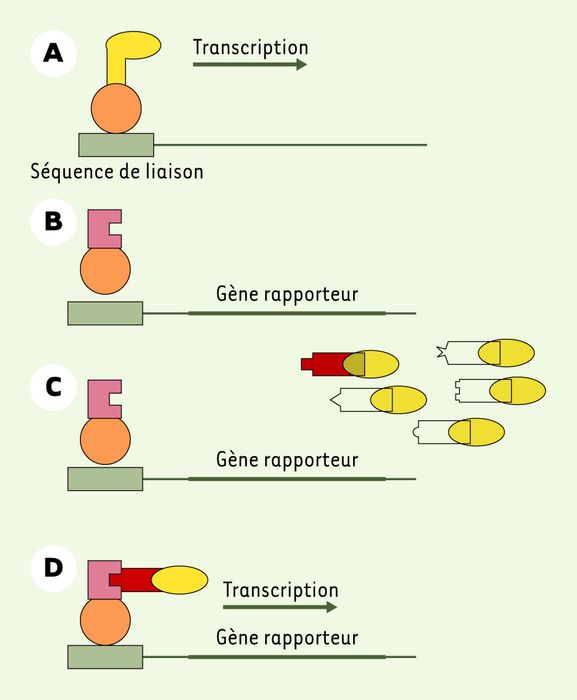
A. La protéine GAL4 de levure est constituée d’un domaine de liaison de l’ADN (orange) et d’un domaine d’activation de la transcription (jaune). B. Dans l’approche double-hybride, une construction de sélection contenant un gène rapporteur sous le contrôle de la séquence de liaison de GAL4 est introduite, ainsi qu’une fusion entre le domaine de liaison de GAL4 et la protéine d’intérêt (l’« appât », en rose). C. Ces levures modifiées sont ensuite transfectées par une banque d’ADNc préparée à partir du tissu d’intérêt, dans laquelle les ADNc sont fusionnés avec la séquence codant pour le domaine de transactivation. D. Si une protéine codée par cette banque d’ADNc complémentaire (la « proie », en rouge) interagit avec la protéine « appât », une reconstitution fonctionnelle a lieu et permet l’expression du gène rapporteur (transcription).
La technique TAP-TAG
Ces limites n’existent pas dans l’autre technique utilisée en protéomique d’interaction, le TAP-TAG (tandem affinity purification by tag) (Figure 5) [16]. Dans cette approche, ce sont les complexes présents dans la cellule d’intérêt qui sont extraits puis analysés. De ce fait, l’influence des modifications post-traductionnelles est prise en compte. En revanche, la difficulté de cette approche est de deux ordres. Le premier est de diminuer les interactions non spécifiques. Ce problème majeur des techniques d’affinité (immunoprécipitation par exemple) a été résolu par la structure de l’étiquette d’affinité et le schéma de purification en tandem [16]. En revanche, le deuxième problème, celui de l’extraction des complexes, reste non résolu. En effet, des interactions protéine-protéines labiles, qui sont détectables dans la cellule par la technique du double hybride, peuvent ne pas survivre au processus d’extraction utilisé. A contrario, le processus de lyse cellulaire peut aussi induire des interactions artéfactuelles. Cependant, cette technique permet de déterminer des complexes protéiques à l’échelle de cellules entières [17].
Figure 5
Analyse de complexes par une méthode de type TAPTAG (tandem affinity purification by tag).

A. La cellule d’intérêt est transfectée avec une construction codant pour la protéine « appât » (en rose), dont on veut déterminer les partenaires, fusionnée avec le module de purification (en noir). B. Le complexe bâti autour de la protéine « appât » est extrait de la cellule, puis (C) purifié par chromatographie d’affinité (en deux étapes). D. Le complexe élué est ensuite analysé par analyse protéomique.
Du fait des biais différents présentés par ces deux techniques, leur complémentarité apparaît évidente [18-20].
Applications thérapeutiques de l’analyse protéomique
Du fait de la redoutable complexité du monde des protéines, les techniques d’analyse protéomique, que celle-ci soit d’expression ou d’interaction, ne peuvent pas relever le défi de l’exhaustivité. En revanche, comme le niveau d’analyse est très proche de la réalité de la physiologie cellulaire, des analyses relativement partielles peuvent donner des résultats pertinents. Dans le cadre de cet article, l’accent sera mis sur des applications potentielles en thérapeutique.
Recherche de marqueurs diagnostiques
Un des domaines où l’analyse protéomique présente un fort potentiel est celui des marqueurs de diagnostic et de pronostic. En effet, des marqueurs secondaires peuvent avoir un très bon potentiel prédictif. En conséquence, les limitations de la protéomique d’expression dans l’analyse des protéines mineures ne sont pas rédhibitoires. Un exemple de recherche de marqueur diagnostique par analyse protéomique est celui d’une mise en évidence, dans le liquide céphalorachidien, de la protéine 14-3-3 σ comme marqueur de la maladie de Creutzfeldt-Jakob [21]. Cette protéine est en fait une protéine abondante dans le cerveau, et sa présence en forte quantité dans le liquide céphalorachidien signe une destruction cérébrale d’un type particulier, caractéristique de la maladie de Creutzfeldt-Jakob et de l’encéphalite herpétique.
Compréhension moléculaire des maladies
Du fait de sa généralité, l’approche protéomique peut être utilisée pour augmenter la compréhension des maladies en vue d’un meilleur traitement ou, au moins, d’un meilleur diagnostic. Dans ce cadre, le fait que l’analyse protéomique ne suppose pas d’hypothèses mécanistiques permet une approche sans a priori. En revanche, de nombreuses maladies font intervenir des protéines faiblement abondantes (par exemple les proto- ou anti-oncogènes) ou membranaires (comme dans la mucoviscidose), souvent difficiles à mettre en évidence. De plus, des mutations ponctuelles, difficiles à détecter en analyse protéomique, peuvent jouer un rôle crucial dans l’étiologie de certains cancers.
Ces limites de l’analyse protéomique constituent un obstacle à la détection de cibles primaires susceptibles de donner lieu à des développements médicamenteux. En revanche, l’analyse protéomique est souvent capable de fournir une description moléculaire phénotypique des maladies. L’étude des phénomènes de transdifférenciation épithéliale dans certains cancers de la vessie en est un excellent exemple [22]. Dans cette étude, l’analyse protéomique a permis de mettre en évidence une expression déréglée de certaines isoformes de kératines, véritables marqueurs pouvant ensuite être étudiés en détail par des techniques immunocytochimiques, par exemple pour une délimitation précise de la tumeur au sein du tissu.
L’étude des maladies virales peut également bénéficier des apports de l’analyse protéomique. Cette approche est en particulier bien adaptée à l’étude des mécanismes mis en place par les virus pour détourner la machinerie cellulaire à leur profit [23]. Au-delà de la compréhension fondamentale, ce type d’étude pourrait permettre d’identifier des cibles cellulaires susceptibles d’être utilisées pour un traitement des affections virales.
Compréhension moléculaire des relations hôte-pathogène
Un cas particulier est représenté par l’étude des agents pathogènes et de la réponse immunitaire induite. Dans ce cadre, les études protéomiques sont particulièrement attractives dans plusieurs domaines. L’un d’entre eux est représenté par l’étude comparative des protéomes d’agents infectieux, en particulier bactériens ou parasitaires, pour identifier des corrélations entre protéome et virulence [24], permettant par la suite de définir des cibles thérapeutiques potentielles.
L’étude de la réponse immune induite au cours des infections est également un domaine où l’apport de l’analyse protéomique est intéressant. Il est ainsi possible de déterminer, par une approche protéomique, la nature des protéines de l’agent pathogène induisant une réponse immunitaire [25, 26]. Ces protéines représentent ensuite des cibles de choix pour le développement de stratégies vaccinales [27]. Il est évident que, dans de telles études, la représentativité de l’étude protéomique constitue un facteur clé de pertinence. Dans le cas des protéomes bactériens, leur relative simplicité favorise les études les plus complètes par les techniques classiques d’analyse protéomique [28].
Études toxicologiques
Le type d’étude mentionné précédemment s’intéresse à la partie la plus en amont de la recherche pharmaceutique, qui va de la description de la maladie à la conception des molécules bioactives. L’analyse protéomique peut également être utilisée avec profit dans la partie située en aval, en particulier pour les études toxicologiques visant à comprendre les effets secondaires des molécules bioactives. Dans ce cadre, l’absence d’hypothèse préconçue et le spectre relativement étendu de l’analyse protéomique sont des avantages certains. Un bon exemple est fourni par l’étude des effets secondaires de la ciclosporine A [29], où l’analyse protéomique a permis de découvrir des aspects mécanistiques de la néphrotoxicité de ce médicament. En revanche, ces analyses toxicologiques supposent la réalisation de grandes séries expérimentales, ce qui nécessite une analyse informatisée des résultats et fait ressentir la faiblesse des outils de bioinformatique.
Conclusions et perspectives
Dans le champ d’application de la recherche thérapeutique, les limitations technologiques de l’analyse protéomique se font clairement sentir. Outre les difficultés liées à la gestion de très grands ensembles de données, communes à toutes les analyses à grande échelle, la protéomique d’expression fait ressortir des obstacles liés à l‘hétérogénéité des protéines, sur le plan physicochimique comme sur le plan de la dynamique d’expression. En effet, le rapport quantitatif entre les protéines les moins abondantes et les plus abondantes dans une cellule dépasse 106, et atteint 1012 dans le sérum. Si le défi analytique reste immense, il est évident que tout progrès permettant d’accroître le spectre d’analyse aura des retentissements immédiats sur les performances de l’analyse protéomique, que ce soit en recherche à visée fondamentale ou thérapeutique.
Par ailleurs, il devrait être particulièrement intéressant de suivre les applications thérapeutiques de la protéomique d’interaction. S’il devient possible de cribler les diagrammes d’interactions protéine-protéine avec un investissement compatible avec les séries expérimentales importantes requises par la recherche thérapeutique, ce type d’analyse pourrait apporter des informations extrêmement pertinentes dans de nombreux domaines. En effet, s’il a été amplement démontré que les abondances relatives des protéines sont perturbées dans les processus pathologiques, il est légitime de penser que les interactions protéines-protéines peuvent l’être aussi, et être très sensibles à des phénomènes subtils comme des séries de mutations ponctuelles sur plusieurs protéines, difficiles à détecter en protéomique d’expression. La complémentarité entre ces deux branches de l’analyse protéomique apparaît donc évidente. La recherche thérapeutique, qui étudie souvent des phénomènes complexes dans des organismes complexes, devrait être un des principaux bénéficiaires de cette complémentarité. Elle seule permettra de prendre en compte la complexité biologique des protéines, défi bien supérieur à celui représenté par la complexité des génomes.
Appendices
Références
- 1. Anderson NG, Anderson NL. A policy and program for biotechnology. Am Biotechnol Lab 1985 (september-october).
- 2. Fleischmann RD, Adams MD, Whit O, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 1995 ; 269 : 496-512.
- 3. Rabilloud T, Heller M, Gasnier F, et al. Proteomics analysis of cellular response to oxidative stress. Evidence for in vivo overoxidation of peroxiredoxins at their active site. J Biol Chem 2002 ; 277 : 19396-401.
- 4. Wilkins MR, Gasteiger E, Sanchez JC, et al. Two-dimensional gel electrophoresis for proteome projects : the effects of protein hydrophobicity and copy number. Electrophoresis 1998 ; 19 : 1501-5.
- 5. Corthals GL, Wasinger VC, Hochstrasser DF, Sanchez JC. The dynamic range of protein expression : A challenge for proteomic research. Electrophoresis 2000 ; 21 : 1104-15.
- 6. Santoni V, Molloy MP, Rabilloud T. Membrane proteins and proteomics : un amour imposssible ? Electrophoresis 2000 ; 21 : 1054-70.
- 7. Merchant M, Weinberger SR. Recent advancements in surface-enhanced laser desorption/ionization-time of flight-mass spectrometry. Electrophoresis 2000 ; 21 : 1164-77.
- 8. Dare TO, Davies HA, Turton JA, et al. Application of surface-enhanced laser desorption/ionization technology to the detection and identification of urinary parvalbumin-alpha : a biomarker of compound-induced skeletal muscle toxicity in the rat. Electrophoresis 2002 ; 23 : 3241-51.
- 9. Washburn MP, Wolters D, Yates JR 3rd. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol 2001 ; 19 : 242-7.
- 10. MacCoss MJ, McDonald WH, Saraf A, et al. Shotgun identification of protein modifications from protein complexes and lens tissue. Proc Natl Acad Sci USA 2002 ; 99 : 7900-5.
- 11. Gygi SP, Rist B, Gerber SA, et al. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol 1999 ; 17 : 994-9.
- 12. Han DK, Eng J, Zhou H, Aebersold R. Quantitative profiling of differentiation-induced microsomal proteins using isotope-coded affinity tags and mass spectrometry. Nat Biotechnol 2001 ; 19 : 946-51.
- 13. Koller A, Washburn MP, Lange BM, et al. Proteomic survey of metabolic pathways in rice. Proc Natl Acad Sci USA 2002 ; 99 : 11969-74.
- 14. Fields S, Song OK. A novel genetic system to detect protein-protein interactions. Nature 1989 ; 340 : 245-6.
- 15. Rain JC, Selig L, De Reuse H, et al. The protein-protein interaction map of Helicobacter pylori. Nature 2001 ; 409 : 211-5.
- 16. Puig O, Caspary F, Rigaut G, et al. The tandem affinity purification (TAP) method : A general procedure of protein complex purification. Methods 2001 ; 24 : 218-29.
- 17. Gavin AC, Bosche M, Krause R, et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 2002 ; 415 : 141-7.
- 18. Bouveret E, Rigaut G, Shevchenko A, et al. A Sm-like protein complex that participates in mRNA degradation. EMBO J 2000 ; 19 : 1661-71.
- 19. Fromont-Racine M, Mayes AE, Brunet-Simon A, et al. Genome-wide protein interaction screens reveal functional networks involving Sm-like proteins. Yeast 2000 ; 17 : 95-110.
- 20. Bader GD, Hogue CW. Analyzing yeast protein-protein interaction data obtained from different sources. Nat Biotechnol 2002 ; 20 : 991-7.
- 21. Hsich G, Kenney K, Gibbs CJ, et al. The 14-3-3 brain protein in cerebrospinal fluid as a marker for transmissible spongiform encephalopathies. N Engl J Med 1996 ; 335 : 924-30.
- 22. Celis JE, Wolf H, Ostergaard M. Bladder squamous cell carcinoma biomarkers derived from proteomics. Electrophoresis 2000 ; 21 : 2115-21.
- 23. Greco A, Bausch N, Coute Y, Diaz JJ. Characterization by two-dimensional gel electrophoresis of host proteins whose synthesis is sustained or stimulated during the course of Herpes simplex virus type 1 infection. Electrophoresis 2000 ; 21 : 2522-30.
- 24. Deiwick J, Rappl C, Stender S, et al. Proteomic approaches to Salmonella pathogenicity Island 2 encoded proteins and the SsrAB regulon. Proteomics 2002 ; 2 : 792-9.
- 25. Sanchez-Campillo M, Bini L, Comanducci M, et al. Identification of immunoreactive proteins of Chlamydia trachomatis by Western blot analysis of a two-dimensional electrophoresis map with patient sera. Electrophoresis 1999 ; 20 : 2269-79.
- 26. Nilsson CL, Larsson T, Gustafsson E, et al. Identification of protein vaccine candidates from Helicobacter pylori using a preparative two-dimensional electrophoretic procedure and mass spectrometry. Anal Chem 2000 ; 72 : 2148-53.
- 27. Grandi G. Antibacterial vaccine design using genomics and proteomics. Trends Biotechnol 2001 ; 19 : 181-8.
- 28. Tonella L, Hoogland C, Binz PA, et al. New perspectives in the Escherichia coli proteome investigation. Proteomics 2001 ; 1 : 409-23.
- 29. Aicher L, Wahl D, Arce A, et al. New insights into cyclosporine A nephrotoxicity by proteome analysis. Electrophoresis 1998 ; 19 : 1998-2003.
List of figures
Figure 1
Analyse d’un mélange de protéines par électrophorèse bidimensionnelle et spectrométrie de masse.
A. Le mélange de protéines est séparé par électrophorèse bidimensionnelle. B. Les taches d’intérêt, observées après coloration, sont digérées pour donner des peptides. C. Ces peptides sont extraits puis analysés par spectromètre de masse. Cette analyse fournit le nom des protéines présentes dans le fragment de gel de départ, ainsi que certaines des modifications post-traductionnelles portées par cette forme protéique.
Figure 2
Analyse d’un mélange de protéines par la méthode MUDPIT (multidimensional protein identification technology).
A. Le mélange de protéines est directement digéré par la trypsine. B. Le mélange complexe de peptides résultant de cette digestion est ensuite acidifié avec de l’acide acétique (AcOH). C. Le mélange est déposé sur un système composé de deux colonnes chromatographiques en tandem, relié à un spectromètre de masse (MS) en tandem : la première colonne est un échangeur fort de cations (CE), la deuxième une phase inverse (PI). D. L’élution de la colonne échangeuse d’ions par une concentration donnée de sels (NH4HCO3) transfère une partie des peptides vers la colonne de phase inverse. E. Après lavage pour éliminer le sel, la colonne de phase inverse est développée par un gradient d’éthanol (EtOH, 1 heure), et les peptides sortant de la colonne sont analysés par spectrométrie de masse en tandem, ce qui permet de déterminer leur nature, et donc la protéine dont ils proviennent. Une fois l’élution terminée, une étape de salinité plus importante est appliquée pour transférer d’autres peptides vers la colonne de phase inverse ; une vingtaine de ces cycles sont réalisés.
Figure 3
Analyse comparative par la méthode ICAT (isotope coded affinity tag).
A. Schéma d’une sonde ICAT spécifique des cystéines. Cette sonde est constituée de trois parties : une partie réagissant avec les cystéines (iodoacétamide), une biotine (à gauche) qui permet la sélection par chromatographie d’affinité et un bras espaceur qui peut contenir des hydrogènes (carrés) ou des deutérium (ronds). B. Les deux extraits à comparer sont ensuite marqués, l’un avec la sonde légère (hydrogène), l’autre avec la sonde lourde (deutérium). C. Les extraits sont mélangés et digérés par la trypsine, puis les peptides marqués par la sonde sont sélectionnés par chromatographie d’affinité. D. Les peptides sélectionnés sont analysés par chromatographie liquide (phase inverse) (CPL/PI) couplée en ligne avec un spectromètre de masse (MS) en tandem. Le rapport des signaux correspondant aux peptides identiques marqués par la sonde légère et la sonde lourde, observé dans le premier analyseur du spectromètre de masse, donne le rapport quantitatif des protéines d’origine. Les données de fragmentation des peptides donnent la nature de la protéine d’origine. On notera que les protéines dénuées de cystéines échappent à l’analyse.
Figure 4
L’approche double-hybride.
A. La protéine GAL4 de levure est constituée d’un domaine de liaison de l’ADN (orange) et d’un domaine d’activation de la transcription (jaune). B. Dans l’approche double-hybride, une construction de sélection contenant un gène rapporteur sous le contrôle de la séquence de liaison de GAL4 est introduite, ainsi qu’une fusion entre le domaine de liaison de GAL4 et la protéine d’intérêt (l’« appât », en rose). C. Ces levures modifiées sont ensuite transfectées par une banque d’ADNc préparée à partir du tissu d’intérêt, dans laquelle les ADNc sont fusionnés avec la séquence codant pour le domaine de transactivation. D. Si une protéine codée par cette banque d’ADNc complémentaire (la « proie », en rouge) interagit avec la protéine « appât », une reconstitution fonctionnelle a lieu et permet l’expression du gène rapporteur (transcription).
Figure 5
Analyse de complexes par une méthode de type TAPTAG (tandem affinity purification by tag).
A. La cellule d’intérêt est transfectée avec une construction codant pour la protéine « appât » (en rose), dont on veut déterminer les partenaires, fusionnée avec le module de purification (en noir). B. Le complexe bâti autour de la protéine « appât » est extrait de la cellule, puis (C) purifié par chromatographie d’affinité (en deux étapes). D. Le complexe élué est ensuite analysé par analyse protéomique.