Article body
Les mécanismes de tolérance immunitaire sont destinés à prévenir le développement des maladies auto-immunes. Concernant les lymphocytes B (LyB), les processus d’élimination décrits à ce jour (délétion clonale et anergie qui aboutissent à l’élimination de la cellule et édition du récepteur qui conduit à une perte de l’autoréactivité) ne sont que partiellement efficaces [1]. En effet, les sujets sains possèdent de nombreux LyB autoréactifs. Dans les conditions physiologiques, ils ne sont pas activés et coexistent « en paix » avec leur auto-antigène. Les mécanismes que nous-mêmes et d’autres avons proposés d’appeler « ignorance immunologique » sont imparfaitement élucidés [2]. La rupture de cet état est certainement un des mécanismes à l’origine des maladies auto-immunes. De nombreux facteurs génétiques influençant la susceptibilité aux maladies auto-immunes ont été identifiés. Néanmoins, ils apparaissent généralement insuffisants, suggérant un rôle important pour des facteurs environnementaux. Parmi ceux-ci, les infections jouent très probablement un rôle, suspecté de longue date, bien que les mécanismes physiopathologiques en restent mal connus [3]. Une hypothèse (la théorie dite antigène non-spécifique) formule que les dégâts tissulaires infligés par l’infection microbienne pourraient exposer des auto-antigènes aux lymphocytes autoréactifs.
Nous proposons un modèle mécanistique plus précis et original dans lequel l’infection par une bactérie induit in vivo la production d’auto-anticorps : la stimulation simultanée de récepteurs Toll-like (TLR) et du récepteur de l’antigène des LyB (BCR) à la surface de lymphocytes autoréactifs induit une rupture de leur « ignorance immunologique » [4]. Les TLR sont les principaux récepteurs de l’immunité innée décrits à ce jour chez les mammifères [5]. Ils reconnaissent des motifs biochimiques conservés particuliers aux bactéries et aux virus appelés PAMP (pathogen-associated molecular patterns). Les TLR sont exprimés par les cellules du système immunitaire et jouent un rôle important dans l’initiation des réponses immunitaires adaptatives en permettant notamment l’activation des cellules présentatrices d’antigènes (cellules dendritiques et macrophages). Par exemple, TLR4 reconnaît le lipopolysaccharide qui est un composant de la membrane externe des bactéries Gram- tandis que TLR2 reconnait les lipoprotéines de certaines bactéries telles que Borrelia burgdorferi, l’agent de la maladie de Lyme [6].
Nous avons établi un modèle de souris transgéniques (tg) qui reproduit l’état d’« ignorance immunologique » physiologique [2, 4, 7]. Chez ces animaux, la plupart des LyB expriment un facteur rhumatoïde (FR) humain, soit de faible affinité (lignée Smi), soit de forte affinité (lignée Hul). Les FR sont des auto-anticorps reconnaissant la région constante des immunoglobulines G (IgG) et qui apparaissent dans le sérum au cours de nombreuses maladies auto-immunes telles que la polyar-thrite rhumatoïde ou le lupus. Chez le sujet sain, les LyB FR sont présents en nombre élevé mais ne produisent quasiment pas de FR malgré la présence de quantités importantes d’IgG. Ils sont typiquement « ignorants » ; leur rôle physiologique reste mystérieux [8]. Les FR Smi et Hul ne reconnaissant pas les IgG de souris, les LyB FR ne sont pas en situation d’autoréactivité chez les animaux tg. En revanche, lorsque l’on croise des souris tg Smi ou Hul avec des souris ayant un knock-in pour le gène de la région constante des IgG1 humaines (souris SmixcIgG ou HulxcIgG) et qui produisent des IgG chimériques (cIgG) à régions constantes humaines et régions variables murines, les LyB FR tg sont placés en situation d’autoréactivité (Figure 1). Sur un fond génétique non connu pour favoriser l’auto-immunité, ils coexistent en paix avec les cIgG et ne produisent que très peu de FR dans le sérum (situation identique à celle de l’homme sain). L’infection des souris tg par Borrelia burgdorferi entraîne une activation et une prolifération des LyB FR dans les ganglions ainsi qu’une production de FR dans le sérum. L’activation des LyB est en partie non-spécifique, liée à l’engagement de TLR puisqu’elle est abolie en l’absence de MyD88 (principal adaptateur de la voie de signalisation des TLR). Mais le résultat le plus intéressant est que la production de FR n’est significative et prolongée que lorsque l’auto-antigène (les cIgG) est présent et que l’affinité du FR est élevée (c’est-à dire chez les animaux Hul x cIgG). In vitro, les LyB FR ne sont pas activables par les IgG humaines seules, le sont modérément par des extraits de Borrelia, mais sont fortement activés par des complexes immuns IgG-Borrelia. Cette suractivation est bloquée par la ciclosporine A, connue pour prévenir l’activation des LyB induite par le BCR, sans interférer avec la voie TLR [9]. Nous proposons qu’existe une synergie entre les voies TLR et BCR induites par les complexes immuns IgG-Borrelia. En d’autres termes, l’activation de la voie TLR rend les LyB, auparavant « ignorants », activables par leur auto-antigène. Cette co-signalisation induit l’activation et la prolifération des LyB, mais la pleine production de FR reste dépendante de la présence de LyT CD4, comme l’attestent des expériences in vivo où l’administration d’un anticorps anti-CD4 bloquant prévient la surproduction de FR dans les animaux Hul x cIgG infectés. Or, les LyB FR ne peuvent recevoir l’aide de LyT anti-cIgG car ils sont éliminés par les mécanismes de tolérance. Le scénario le plus probable est que les LyB FR, stimulés par les complexes immuns IgG-Borrelia, sont capables, après les avoir internalisés, de recevoir l’aide de LyT spécifiques en leur présentant des antigènes borréliens (Figure 2) [10].
Figure 1
Production des souris Hul x cIgG.
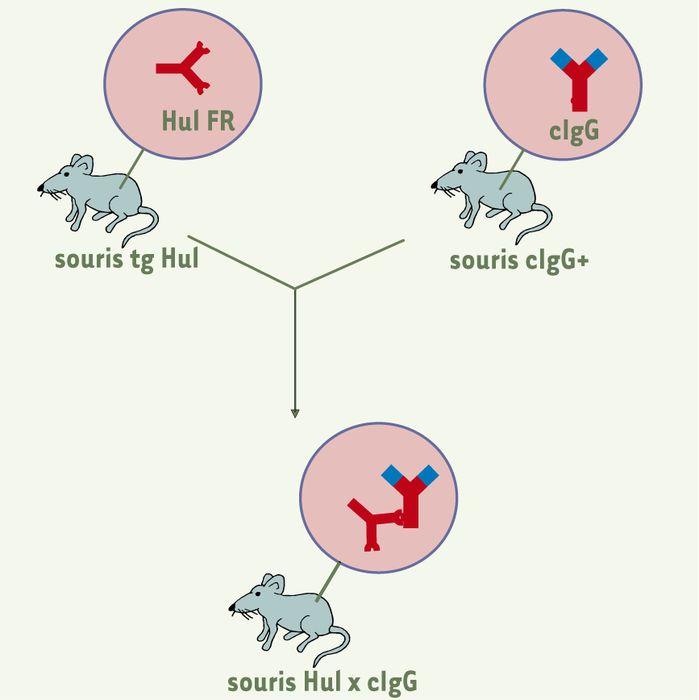
Les souris transgéniques (tg) pour le facteur rhumatoïde (FR) humain Hul sont croisées avec des souris knock-in chez lesquelles le gène codant pour la région constante des IgG1 à été remplacé par son équivalent humain. Les souris cIgG+ produisent des IgG chimériques à régions variables murines et à région constante humaine ; à l’inverse des IgG murines, les cIgG sont reconnues par les FR humains. Les régions en rouge sont d’origine humaine, celles en bleu sont murines. Les souris Smi x cIgG sont obtenues de manière analogue.
Figure 2
Mécanisme possible d’activation des LyB exprimant des FR Hul dans les souris Hul x cIgG par des complexes immuns contenant des IgG anti-B. burgdorferi (Bb) et des antigènes de B. burgdorferi.
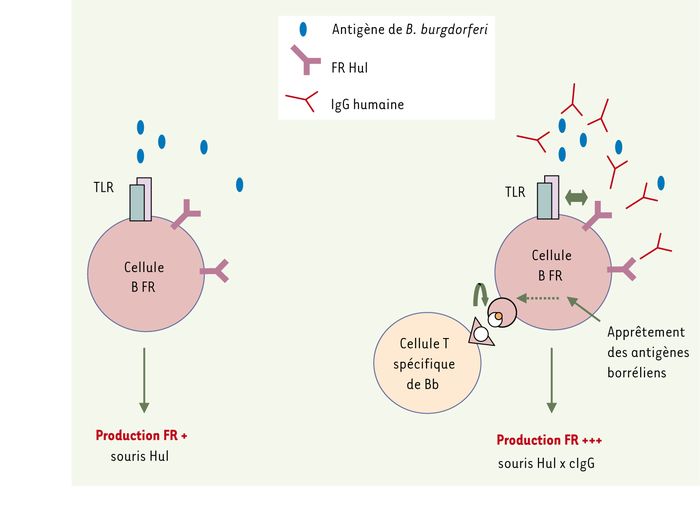
A. Les LyB, y compris les LyB FR, sont stimulés par B. burgdorferi par l’intermédiaire d’une interaction avec des TLR dont TLR2. Dans les souris Hul, le transgène FR ne reconnaissant rien, il n’y a pas de signal par le BCR. B. la présence de cIgG anti-B. burgdorferi dans les souris Hul x cIgG conduit à la formation de complexes immuns contenant B. burgdorferi qui induisent un co-signal par l’intermédiaire de TLR et du BCR de spécificité FR. Les antigènes de B. burgdorferi après internalisation dans les LyB FR par le BCR sont apprêtés en peptides ; des LyT spécifiques de B. burgdorferi coopèrent pour augmenter l’activation des LyB FR et la production de FR en « croyant » aider des LyB anti-B. burgdorferi (+ et +++ représentent l’intensité de production de FR) (adapté d’après [4] avec la permission de J Clin Invest).
Ce travail, soulignant l’implication possible des TLR dans l’initiation de l’auto-immunité, devrait ouvrir de nouvelles perspectives thérapeutiques. Il reste à présent à comprendre pourquoi tous les sujets ne développent pas une pathologie auto-immune au décours d’un état infectieux.
Appendices
Références
- 1. Goodnow CC, Sprent J, Fazekas de St Groth B, Vinuesa CG. Cellular and genetic mechanisms of self tolerance and autoimmunity. Nature 2005 ; 435 : 590-7.
- 2. Koenig-Marrony S, Soulas P, Julien S, et al. Natural autoreactive B cells in transgenic mice reproduce an apparent paradox to the clonal tolerance theory. J Immunol 2001 ; 166 : 1463-70.
- 3. Christen U, Von Herrath MG. Infections and autoimmunity-good or bad ? J Immunol 2005 ; 174 : 7481-6.
- 4. Soulas P, Woods A, Jaulhac B, et al. Autoantigen, innate immunity, and T cells cooperate to break B cell tolerance during bacterial infection. J Clin Invest 2005 ; 115 : 2257-67.
- 5. Akira S, Takeda K. Toll-like receptor signalling. Nat Rev 2004 ; 4 : 499-511.
- 6. Alexopoulou L, Thomas V, Schnare M, et al. Hyporesponsiveness to vaccination with Borrelia burgdorferi OspA in humans and in TLR1- and TLR2- deficient mice. Nat Med 2002 ; 8 : 878-84.
- 7. Julien S, Soulas P, Garaud JC, et al. B cell positive selection by soluble self-antigen. J Immunol 2002 ; 169 : 4198-204.
- 8. Dorner T, Egerer K, Feist E, Burmester GR. Rheumatoid factor revisited. Curr Opin Rheumatol 2004 ; 16 : 246-53.
- 9. Rui L, Vinuesa CG, Blasioli J, Goodnow CC. Resistance to CpG DNA-induced autoimmunity through tolerogenic B cell antigen receptor ERK signaling. Nat Immunol 2003 ; 4 : 594-600.
- 10. Roosnek E, Lanzavecchia, A. Efficient and selective presentation of antigen-antibody complexes by rheumatoid factor B cells. J Exp Med 1991 ; 173 : 487-9.
List of figures
Figure 1
Production des souris Hul x cIgG.
Les souris transgéniques (tg) pour le facteur rhumatoïde (FR) humain Hul sont croisées avec des souris knock-in chez lesquelles le gène codant pour la région constante des IgG1 à été remplacé par son équivalent humain. Les souris cIgG+ produisent des IgG chimériques à régions variables murines et à région constante humaine ; à l’inverse des IgG murines, les cIgG sont reconnues par les FR humains. Les régions en rouge sont d’origine humaine, celles en bleu sont murines. Les souris Smi x cIgG sont obtenues de manière analogue.
Figure 2
Mécanisme possible d’activation des LyB exprimant des FR Hul dans les souris Hul x cIgG par des complexes immuns contenant des IgG anti-B. burgdorferi (Bb) et des antigènes de B. burgdorferi.
A. Les LyB, y compris les LyB FR, sont stimulés par B. burgdorferi par l’intermédiaire d’une interaction avec des TLR dont TLR2. Dans les souris Hul, le transgène FR ne reconnaissant rien, il n’y a pas de signal par le BCR. B. la présence de cIgG anti-B. burgdorferi dans les souris Hul x cIgG conduit à la formation de complexes immuns contenant B. burgdorferi qui induisent un co-signal par l’intermédiaire de TLR et du BCR de spécificité FR. Les antigènes de B. burgdorferi après internalisation dans les LyB FR par le BCR sont apprêtés en peptides ; des LyT spécifiques de B. burgdorferi coopèrent pour augmenter l’activation des LyB FR et la production de FR en « croyant » aider des LyB anti-B. burgdorferi (+ et +++ représentent l’intensité de production de FR) (adapté d’après [4] avec la permission de J Clin Invest).