Article body
Les membres de la famille du TNF (tumor necrosis factor) jouent un rôle dans la prolifération, la différenciation et la mort cellulaires, notamment dans le contexte de la réponse immunitaire et de la réaction inflammatoire [1]. Certains récepteurs de ces cytokines possèdent en commun un motif intracellulaire très conservé, constitué d’une soixantaine d’acides aminés, nommé domaine de mort (death domain). Ce motif est nécessaire à l’activation du processus de mort cellulaire en réponse à l’engagement du récepteur correspondant. Par exemple, en réponse à l’interaction du ligand de Fas (CD95-L) avec son récepteur (Fas/CD95) ou celle de TRAIL avec l’un de ses récepteurs, ce motif interagit avec la molécule adaptatrice FADD (Fas-associated death domain) par l’intermédiaire de laquelle le récepteur recrute la procaspase-8. Du fait de la trimérisation du récepteur, nécessaire à son interaction avec le ligand, le recrutement de la procaspase-8 par chaque récepteur permet un enrichissement local en cette enzyme qui favorise son autoactivation. L’activation de la caspase-8 est la première étape d’une cascade protéolytique conduisant à la mort cellulaire par apoptose [2].
La voie de signalisation aboutissant à la mort cellulaire en réponse au TNF est plus complexe. Le récepteur 1 du TNF (TNF-R1) possède un domaine de mort comparable à celui de Fas ou des récepteurs de TRAIL et les protéines FADD et caspase-8 sont indispensables au déclenchement de l’apoptose en réponse à l’engagement de ce récepteur [1]. Cependant, contrairement à ce qui est observé en réponse à l’engagement de Fas ou des récepteurs de TRAIL [2], ni FADD, ni la procaspase-8 ne sont recrutés au niveau membranaire lors de l’engagement de TNF-R1 [2]. Cet engagement peut induire des signaux de survie aussi bien que des signaux de mort et nous avons montré récemment que ces effets contradictoires mettaient en jeu deux complexes distincts [3]. La formation d’un premier complexe (complexe I), associé à la membrane plasmique, est responsable de l’activation de la voie de survie impliquant NF-κB. C’est un deuxième complexe (complexe II), cytosolique, qui active les voies de mort cellulaire. Le dialogue entre ces 2 complexes détermine la survie cellulaire (Figure 1).
Figure 1
Modèle d’activation des récepteurs de la famille du TNF (tumor necrosis factor).
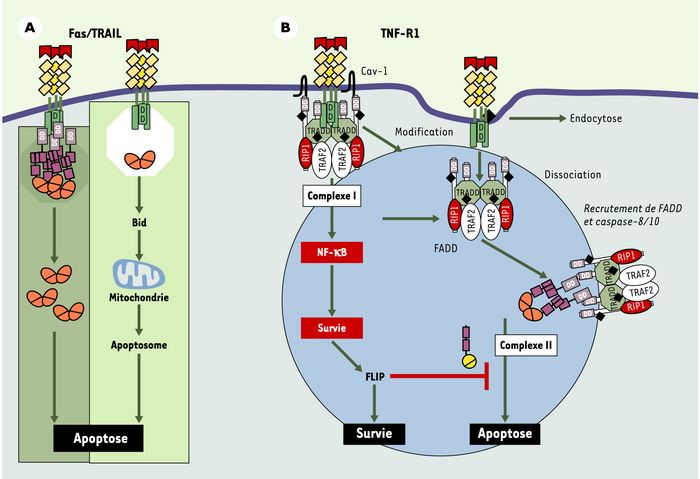
A. Fas et TRAIL induisent l’activation de l’apoptose au niveau de la membrane plasmique via un complexe unique appelé DISC (death inducing signaling complex) dont l’activité protéolytique nécessite parfois une boucle d’amplification du signal impliquant Bid et la mitochondrie (mt). B. L’activation de l’apoptose par TNF-R1 requiert la formation de deux complexes distincts. Le complexe (I), membranaire, est responsable de l’activation de la voie de survie induite par NF-κB. Le complexe (II), pro-apoptotique, se forme dans un second temps, après modification du complexe (I) et dissociation de ces composants du récepteur du TNF, permettant le recrutement de la protéine adaptatrice FADD et des caspases activatrices (-8 et –10). Ce complexe, cytosolique, équivaut au DISC de Fas ou de TRAIL et est capable d’activer l’apoptose lorsque la voie NF-κB est inactive. Dans le cas contraire, la molécule anti-apoptotique, FLIP, réglée positivement par le complexe (I), bloque l’activation de l’apoptose.
Le complexe I associe la molécule adaptatrice TRADD, la kinase RIP, la molécule adaptatrice TRAF-2, et le complexe IKK (IKKα, IKKβ et IKKγ) responsable de l’activation de la voie NF-κB. Au cours de la formation de ce complexe, certaines des protéines qui le constituent sont modifiées par ubiquitinylation dans les «radeaux lipidiques» de la membrane plasmique. L’altération de ces structures riches en lipides prévient la phosphorylation de la protéine I-κBα, et sensibilise les cellules à l’activité pro-apoptotique du TNF [4]. Ni FADD, ni la procaspase-8 ne participent à la formation du complexe I [3, 5], quelle que soit la sensibilité des cellules à l’apoptose induite par le TNF.
En activant NF-κB, le complexe I augmente l’expression de gènes tels que TRAF-1, TRAF-2, c-IAP1 et c-IAP2 [6]. Cependant, nous avons montré qu’aucun de ces gènes ne peut, à lui seul, bloquer l’activité cytotoxique du TNF. En revanche, une autre cible de NF-κB, la molécule FLIP, peut à elle seule inhiber complètement l’activité pro-apoptotique du TNF lorsque son expression augmente [7].
Le complexe II est composé de la quasi-totalité du complexe I, à l’exception du récepteur TNF-R1. à la différence du complexe I, il contient aussi la molécule adaptatrice FADD et les caspases initiatrices 8 et 10. Le passage du complexe I au complexe II semble induit par des modifications de RIP et de TRADD exposant le domaine de mort de TRADD, permettant le recrutement de FADD et, par son intermédiaire, des caspases initiatrices. L’inactivation de TRADD chez la souris devrait à cet égard livrer de précieuses informations.
La composition du complexe II varie en fonction de la capacité des cellules à activer NF-κB en réponse à l’engagement du TNFR1. Dans les cellules dans lesquelles NF-κB est activé, la cellule surexprime FLIP qui participe à la formation du complexe II et prévient le recrutement de la caspase-10 en s’associant préférentiellement à la caspase-8. Le TNF induit alors un signal de survie et le complexe II ne provoque pas la mort cellulaire. Dans les cellules dans lesquelles NF-κB n’est pas activé en réponse à l’engagement du TNFR1, l’expression de FLIP diminue progressivement, permettant le déclenchement de l’apoptose. Du fait de la complexité de cette voie de signalisation, la mort induite par le TNF est plus lente que celle induite par l’engagement de Fas ou des récepteurs de TRAIL. Lorsque la voie NF-κB est fonctionnelle, des inhibiteurs de la traduction tels que le cycloheximide permettent de sensibiliser les cellules au TNF et cela est probablement dû à la diminution de l’expression de FLIP dont la demi-vie est courte.
Ce schéma général complexe (Figure 1) a le mérite de réconcilier l’ensemble des données publiées à l’heure actuelle. Il a cependant été récemment montré que la voie JNK peut également déterminer le devenir des cellules après activation de TNFR1 [8], par un mécanisme post-mitonchondrial impliquant Smac dans le contrôle de l’activation de la pro-caspase-8, notamment en déstabilisant le complexe inhibiteur c-IAP1-TRAF2. Ces données, bien que difficilement conciliables avec le modèle que nous présentons, ouvrent la voie à de nouvelles investigations qui nous permettrons, peut-être un jour, d’élucider le mode opératoire de ce récepteur et de cibler spécifiquement l’un ou l’autre de ces complexes à des fins thérapeutiques visant à traiter les maladies autoimmunes ou certains cancers liés au TNF [9].
Appendices
Références
- 1. Mak TW, Yeh WC. Signaling for survival and apoptosis in the immune system. Arthritis Res 2002; 4 (suppl 3): S243-52.
- 2. Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science 1998; 281: 1305-8.
- 3. Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003; 114: 181-90.
- 4. Legler DF, Micheau O, Doucey MA, Tschopp J, Bron C. Recruitment of TNF receptor 1 to lipid rafts is essential for TNFalpha-mediated NF-kappaB activation. Immunity 2003; 18: 655-64.
- 5. Harper N, Hughes M, MacFarlane M, Cohen GM. Fas-associated death domain protein and caspase-8 are not recruited to the tumor necrosis factor receptor 1 signaling complex during tumor necrosis factor-induced apoptosis. J Biol Chem 2003; 278: 25534-41.
- 6. Wang CY, Mayo MW, Korneluk RG, Goeddel, DV, Baldwin AS Jr. NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science 1998; 281: 1680-3.
- 7. Micheau O, Lens S, Gaide O, Alevizopoulos K, Tschopp J. NF-kappaB signals induce the expression of c-FLIP. Mol Cell Biol 2001; 21: 5299-305.
- 8. Deng Y, Ren X, Yang L, Lin Y, Wu X. A JNK-dependent pathway is required for TNFalpha-induced apoptosis. Cell 2003; 115: 61-70.
- 9. Micheau O. Cellular FLICE-inhibitory protein: an attractive therapeutic target? Expert Opin Ther Targets 2003; 7: 559-73.
List of figures
Figure 1
Modèle d’activation des récepteurs de la famille du TNF (tumor necrosis factor).
A. Fas et TRAIL induisent l’activation de l’apoptose au niveau de la membrane plasmique via un complexe unique appelé DISC (death inducing signaling complex) dont l’activité protéolytique nécessite parfois une boucle d’amplification du signal impliquant Bid et la mitochondrie (mt). B. L’activation de l’apoptose par TNF-R1 requiert la formation de deux complexes distincts. Le complexe (I), membranaire, est responsable de l’activation de la voie de survie induite par NF-κB. Le complexe (II), pro-apoptotique, se forme dans un second temps, après modification du complexe (I) et dissociation de ces composants du récepteur du TNF, permettant le recrutement de la protéine adaptatrice FADD et des caspases activatrices (-8 et –10). Ce complexe, cytosolique, équivaut au DISC de Fas ou de TRAIL et est capable d’activer l’apoptose lorsque la voie NF-κB est inactive. Dans le cas contraire, la molécule anti-apoptotique, FLIP, réglée positivement par le complexe (I), bloque l’activation de l’apoptose.