Abstracts
Résumé
Le développement de cellules cancéreuses est depuis longtemps associé à des dérèglements de l’activité transcriptionnelle de nombreux gènes, mais aussi, plus récemment, à l’apparition d’anomalies d’épissage des ARN prémessagers. La contribution de ces anomalies d’épissage au développement de cellules tumorales, ainsi que le pouvoir tumorigène de certaines des isoformes protéiques qui en sont issues sont encore très peu explorés. Toutefois, depuis la découverte récente du couplage des deux mécanismes - transcription et épissage - des efforts de recherche ont permis de démontrer que des facteurs de transcription oncogéniques affectent aussi l’épissage des prémessagers. Ces observations suscitent des interrogations quant aux mécanismes d’action des oncogènes à activité transcriptionnelle et, à plus long terme, quant à la recherche de cibles cellulaires nouvelles à appréhender dans de futurs protocoles thérapeutiques anticancéreux.
Summary
Oncogene activity ranges from transduction signals to transcription factors. Altered expression of oncogenes, either by chromosomal translocation, proviral insertion or point mutations, can lead to tumor formation. More specifically, data accumulated through the last two decades have shown that disregulation of oncogenic transcription factors can interfere with regulatory cascades that control the growth, differentiation, and survival of normal cells. There is also evidence that alterations of oncogene activity are associated with pre-mRNA splicing defects. The insights gained from the pivotal role of RNA polymerase II in coupling transcription and splicing have instigated a new line of research regarding the possible role of oncogenic transcription factors in pre-mRNA splicing regulation. This review focuses on recent advances addressing this question. Understanding the impact of alterations in the expression and/or function of oncogenes have important prognostic implications that can guide the design of new therapeutic drugs to promote differentiation and/or apoptosis over cell proliferation.
Article body
Un oncogène est défini, au sens large, comme un gène dont un dérèglement de l’expression ou une altération de la structure engendre la croissance incontrôlée de la cellule et, souvent, contribue à l’apparition de cellules tumorales. Les oncogènes codent pour diverses fonctions fondamentales pour la cellule concernant aussi bien le milieu extracellulaire que l’ensemble des compartiments cellulaires. Leurs produits sont aussi bien des facteurs de croissance, des récepteurs de facteurs de croissance, des protéines G de transduction du signal, des protéines à activité tyrosine kinase intracellulaire ou sérine/thréonine kinase, des molécules de signalisation, des facteurs de transcription ou encore des molécules impliquées dans le remodelage de la chromatine [1].
Si le rôle de certains oncogènes en tant que facteurs de transcription a souvent été mis en évidence dans la transformation cellulaire et le développement de cellules cancéreuses [1], quelques travaux, émergeant depuis quelques années, mettent l’accent sur leur rôle possible dans la maturation des ARN messagers, et plus précisément dans l’épissage des pré-messagers.
Dérèglement de la transcription de gènes cibles par la modification de l’activité transcriptionnelle de facteurs proto-oncogènes
En dehors de quelques mutations ponctuelles ou de microdélétions chromosomiques indétectables, deux événements peuvent être à l’origine d’un dérèglement du taux d’expression, et par conséquent de l’activité transcriptionnelle des oncogènes : le premier est l’insertion rétrovirale du génome d’un virus infectant une cellule hôte, le deuxième est une anomalie chromosomique (Figure 1).
Figure 1
Remaniements génomiques conduisant à un dérèglement de l’expression des oncogènes.
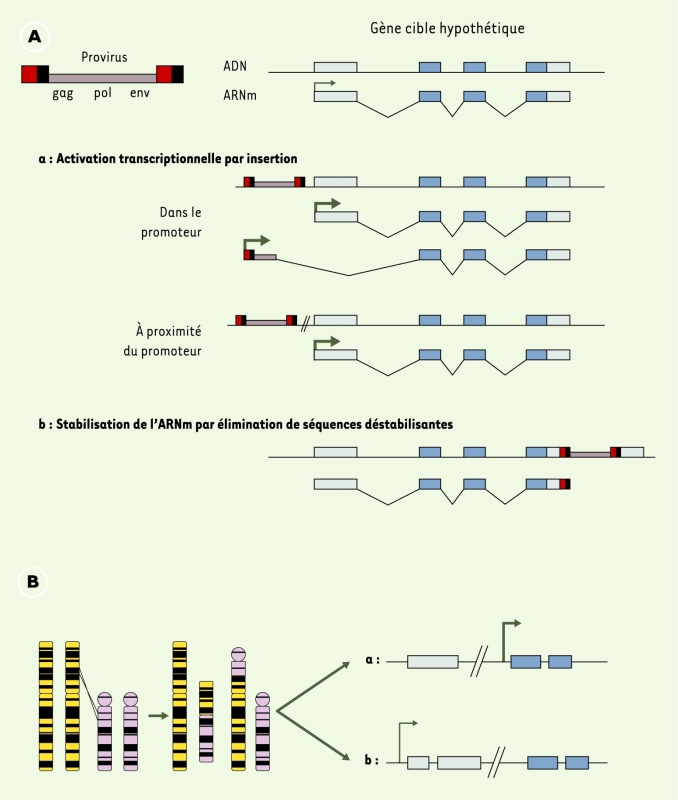
A. Mutagenèse par insertion rétrovirale dans des gènes hôtes. Les rectangles bleus sont les séquences codant pour la protéine finale. L’insertion du provirus aboutit à l’activation de l’expression du gène par augmentation de la quantité d’ARNm disponible pour la traduction, via une activation transcriptionnelle (a) ou une stabilisation du lot d’ARNm à partir d’un promoteur non altéré (b). B. Perturbation du contexte transcriptionnel par translocation chromosomique. Le nouveau contexte chromosomique conduit à l’activation de l’expression du gène, comme pour une insertion rétrovirale (a), ou la translocation conduit à l’apparition d’un gène de fusion entre les exons de deux gènes (b).
Modification de l’activité transcriptionnelle par insertion rétrovirale
Les oncogènes ont été définis au départ comme des gènes viraux induisant la transformation des cellules hôtes. Il est clair aujourd’hui que le pouvoir tumorigène de ces virus est lié à un mécanisme de mutagenèse insertionnelle : l’ADN viral, en s’insérant dans le génome de la cellule hôte, se comporte comme un agent mutagène qui conduit au dérèglement de l’expression d’un gène cellulaire (Figure 1). Le facteur c-myc est probablement l’exemple le mieux connu. Cette molécule-clé règle aussi bien la prolifération que la croissance, la différenciation ou l’apoptose cellulaire via ses activités dans la transcription et le remodelage de la chromatine. Une surexpression de c-myc est retrouvée dans de nombreux types de cancers : lymphomes, leucémies myéloïdes, carcinomes du côlon, du poumon, du sein, des ovaires… [2].
La propriété d’activation de proto-oncogènes par insertion rétrovirale a été récemment utilisée pour développer des techniques d’identification des gènes impliqués dans l’oncogenèse. Ces stratégies consistent à rechercher les sites communs d’insertion (CIS) de provirus en déterminant les séquences adjacentes au provirus [1].
Modification de l’activité transcriptionnelle par remaniement chromosomique
Si 35 % des cellules leucémiques humaines ont un caryotype normal, 65 % d’entre elles présentent un remaniement chromosomique, majoritairement sous la forme d’inversions intrachromosomiques ou de translocations [3]. Les points de cassure chromosomiques seraient localisées à proximité de proto-oncogènes, alors activés en oncogènes par ces remaniements.
Ces remaniements peuvent aboutir à une expression anormale du gène (Figure 1). L’oncogène c-myc, par exemple, a été identifié comme la cible de translocations dans le lymphome de Burkitt. La conséquence de tels réarrangements est la surexpression du gène modifié, conséquence similaire à celle d’une insertion virale. Les translocations chromosomiques peuvent également aboutir à la production de protéines de fusion aux propriétés différentes de celles des deux protéines originales. Les membres de la famille de facteurs de transcription ETS offrent plusieurs exemples de protéines de fusion conduisant à une transformation néoplastique [4] : ainsi, la fusion du domaine carboxyterminal de Tel (translocation ets leukemia) avec la protéine MN1 est associée à des tumeurs myéloïdes humaines. De même, la fusion de son domaine aminoterminal avec la protéine AML1 a été décrite dans 25 % des cas de leucémies lymphoïdes aiguës. Les protéines de fusion EWS-ETS jouent un rôle important dans le développement des tumeurs d’Ewing. Parmi ces protéines de fusion, EWS-Fli-1 et EWS-ERG agissent comme des facteurs de transcription reconnaissant des séquences spécifiques. Leur partie aminoterminale EWS est un transactivateur, et leur partie ETS est un domaine de liaison à l’ADN [4]. Il est probable que leur fonction activatrice de la transcription soit associée à leur potentiel oncogénique. C’est ainsi que l’activité transcriptionnelle et le pouvoir transformant de EWS-Fli sont supérieurs à ceux de Fli-1.
Intervention des facteurs de transcription oncogènes dans l’épissage
Outre des dérèglements de l’expression des gènes au niveau transcriptionnel, plusieurs travaux ont mis l’accent sur des dérèglements qualitatifs de l’information génétique, via des défauts d’épissage des prémessagers, ou via l’apparition de profils d’épissage alternatif spécifiques de certaines tumeurs. Un des exemples les plus étudiés est l’expression différentielle de nombreuses isoformes de CD44 dans diverses tumeurs [5]. Par ailleurs, certaines isoformes issues d’un même gène par épissage alternatif semblent avoir des propriétés plus ou moins métastasiques, et seraient ainsi de bons marqueurs de tumeurs [6].
L’épissage alternatif des prémessagers est un mécanisme majeur de diversification de l’information génétique. Environ 60 % des gènes humains sont concernés par ce mécanisme, dont la régulation dans la cellule normale est contrôlée selon le type cellulaire, le stade de développement et l’environnement cellulaire. Par ailleurs, on estime que 50 % des anomalies génétiques résultent ou produisent des défauts d’épissage. Alors, qu’en est-il du rôle des facteurs de transcription oncogènes dans la régulation de l’épissage ou dans l’apparition pathologique d’isoformes spécifiques de tumeurs ?
Les protéines de fusion EWS/Fli-1 et TLS/ERG
EWS et TLS appartiennent à une même famille de protéines présentant de grandes similitudes. Toutes deux, capables d’interagir avec l’ARN polymérase II, sont des activateurs transcriptionnels. Mais elles interagissent également avec la protéine YB-1, se liant à la polymérase II, pour régler la sélection des sites d’épissage du prémessager E1A adénoviral ; elles sont aussi copurifiées avec les facteurs d’épissage hnRNPA1, hnRNPC1/C2 ou PSF ; enfin, EWS se lie aux composants essentiels de la machinerie d’épissage SF1 et U1C, et TLS interagit avec des facteurs d‘épissage membres de la famille des protéines SR [7-9]. EWS et TLS semblent donc capables de régler la transcription comme l’épissage.
Dans les translocations EWS/Fli-1 et TLS/ERG, les protéines chimériques conservent leur capacité de liaison à la polymérase II via le domaine aminoterminal d’EWS et de TLS, mais elles ne sont plus capables d’interagir avec les nombreux partenaires d’épissage de EWS et TLS, en particulier les protéines SR. C’est sans doute pour cette raison que les protéines de fusion EWS/Fli-1 et TLS/ERG inhibent l’épissage du prémessager E1A dans les cellules HeLa et modifient le profil d’épissage alternatif de l’ARNm CD44 dans des cellules K562 [7, 9-11]. Si le facteur de transcription Fli-1, seul, n’affecte pas l’épissage du messager viral E1A dépendant des protéines SR, il semble s’opposer à l’épissage relayé par hnRNPA1, simulant alors l’action antagoniste de la protéine SR ASF/SF2 : de fait, une forte expression d’hnRNPA1 par rapport au facteur ASF/SF2 favorise la sélection du site 5’ distal, alors qu’une prédominance de ASF/SF2 favorise la sélection du site 5’ proximal, cette régulation différentielle conduisant à la production de quantités différentes d’isoformes du messager viral E1A. En conséquence, la fusion aboutissant à la production de la protéine EWS/Fli-1 peut s’opposer, contrairement à EWS seul, aux changements de profil d’épissage induits par l’expression de hnRNPA1 [10]. Enfin, les deux protéines chimériques EWS/Fli-1 et TLS/ERG, à la différence de EWS et TLS seules (qui sont alors activatrices), peuvent inhiber la sélection par YB1 de sites d’épissage spécifiques du prémessager E1A [12].
Au total, les protéines oncogéniques EWS/Fli-1 et TLS/ERG dérèglent l’expression de gènes cibles en jouant sur leur transcription ou l’épissage alternatif de leurs messagers. Il est probable que la modification de l’activité transcriptionnelle ou la modification du complexe d’épissage par les facteurs oncogéniques contribuent toutes deux à la transformation des cellules.
La protéine Spi-1/PU.1
Tout comme Fli-1, le facteur de transcription Spi-1/PU.1 est également un membre de la famille Ets. Son expression dans les cellules murines érythroleucémiques induites par le virus à réplication défectueuse SFFV est fortement activée après l’insertion provirale dans le locus du gène pu.1 [13]. Cette surexpression de Spi-1/PU.1 est associée à un blocage de la différenciation et à la capacité de proliférer en l’absence d’érythropoïétine. L’apport dans le milieu d’un inducteur chimique induit la différenciation des cellules, avec production d’hémoglobine et inhibition de l’expression de Spi-1/PU.1. Enfin, la diminution d’expression de Spi-1/PU.1 et de Fli-1 est requise pour la différenciation érythroïde terminale, et la surexpression constitutive de l’un ou l’autre de ces oncogènes est suffisante, seule, pour bloquer la différenciation [13, 14].
Les mécanismes moléculaires de ce blocage de la différenciation commencent à être élucidés. Il a été ainsi montré que Spi-1/PU.1, notamment, agit sur le facteur de transcription érythroïde GATA1 par antagonisme croisé (l’un inhibant l’activité de l’autre), et par inhibition de l’acétylation des facteurs de transcription érythroïdes GATA-1, EKLF et NF-E2 (pour revue voir [15]). Ces deux effets inhibiteurs contribueraient aussi bien au blocage de la différenciation qu’à la leucémogenèse.
Des travaux plus récents ont montré que Spi-1/PU.1 affecte également l’épissage [16, 17]. Plus spécifiquement, Spi-1/PU.1 interagit avec la protéine p54nrb, similaire au facteur d’épissage PSF, capable de se lier à l’ARN ; elle inhibe également l’élimination d’un intron placé sur un prémessager artificiel in vitro [16]. Spi-1/PU.1 interagit aussi avec le facteur d’épissage présomptif TLS : en s’opposant à TLS, elle favoriserait dans des cellules intactes la sélection d’un site proximal 5’ d’épissage aux dépens d’un site distal [17]. Dans l’une comme dans l’autre de ces interactions, le mécanisme proposé est une diminution du taux de p54nrb ou TLS via leur séquestration par Spi-1/PU.1.
L’hypothèse d’un effet de Spi-1/PU.1 sur l’épissage a récemment été renforcée par la découverte d’une cible endogène de cet effet : l’inclusion de l’exon 16 sur le messager mature de 4.1R en fin de différenciation érythroïde. La protéine 4.1R est essentielle à l’intégrité du squelette érythrocytaire en renforçant la liaison spectrine/actine. Cette fonction est portée par un domaine codé par l’exon alternatif 16 et l’exon constitutif 17. L’inclusion de l’exon 16 sur le messager 4.1R en fin de différenciation érythrocytaire permet la synthèse d’un domaine fonctionnel de liaison au complexe spectrine/actine, et constitue par conséquent un événement majeur pour la fonction érythrocytaire de 4.1R. Cet événement est corrélé à l’expression de Spi-1/PU.1 dans les cellules érythroleucémiques : élevée dans les cellules en phase proliférative, l’expression de Spi-1/PU.1 diminue fortement au cours de l’induction de la différenciation, en parallèle avec l’augmentation de la reconnaissance de l’exon 16, aussi bien sur le prémessager endogène que sur un prémessager issu d’un minigène transfecté [18, 19] (Figure 2). La seule surexpression de Spi-1/PU.1 est suffisante pour inhiber cet événement d’épissage [19].
Figure 2
Reconnaissance de l’exon 16 alternatif du prémessager 4.1R au cours de la différenciation érythroïde.
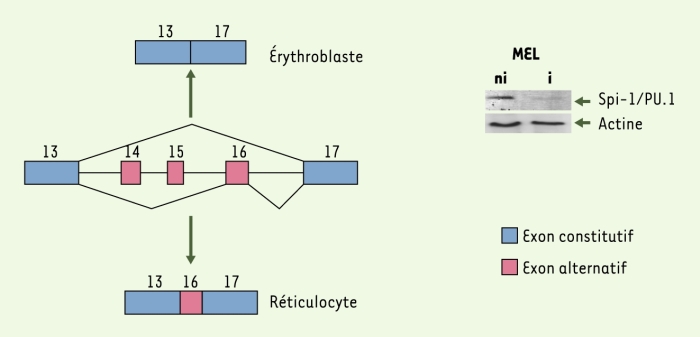
L’exon 16, exclu du messager mature dans les précurseurs, n’est retenu qu’en fin de différenciation érythroïde [18]. Les exons 14 et 15 sont exclus à tous les stades de développement érythroïde et dans la grande majorité des types cellulaires. L’analyse par Western blot utilisant un anticorps anti-Spi-1/PU.1 révèle une expression élevée de la protéine Spi-1/PU.1 dans les cellules érythroleucémiques murines (MEL) non induites (ni), cette expression étant fortement inhibée après induction des cellules (i) vers la différenciation érythroïde terminale (la révélation par un anticorps anti-actine sert de contrôle quantitatif des protéines déposées) [19].
Couplage transcription/épissage
Ces obsevations offrent autant d’exemples de facteurs de transcription oncogéniques fonctionnant à l’interface entre transcription et épissage. Or, il est établi depuis quelques années que transcription et épissage sont des événements cellulaires couplés dans le temps et dans l’espace [20] : l’ARN messager, produit lors de la transcription, est immédiatement pris en charge pour sa maturation : mise en place de la coiffe, épissage et polyadénylation sont ainsi concomitants de la transcription (Figure 3). Il est même démontré que la transcription influe sur la maturation des messagers et vice versa [20]. Ainsi, la structure d’un promoteur et son occupation par des complexes transcriptionnels pourraient modifier l’épissage ; le type de promoteur et la présence d’enhancers sur l’ADN permettraient, de surcroît, de régler le choix du site d’épissage en modifiant la « processivité » de l’ARN polymérase II [21, 22]. Enfin, l’ARN polymérase II, responsable de l’élongation du transcrit, est indispensable au bon déroulement de l’épissage des ARN prémessagers : elle est notamment capable, par son extension carboxyterminale hyperphosphorylée, de recruter directement ou indirectement des facteurs d’épissage.
Figure 3
Transcription et épissage, deux événements couplés pour une meilleure régulation de l’expression des gènes.

Outre la protéine oncogénique Spi-1/PU.1, plusieurs exemples de protéines impliquées dans les deux processus, transcription et épissage, sont déjà connues : P52, un activateur transcriptionnel, est capable d’interagir avec le facteur d’épissage ASF/SF2 [25]. U1 snRNP, facteur d’épissage essentiel du splicéosome, peut se lier à plusieurs protéines impliquées dans la transcription : le facteur d’élongation TAT-SF1 [26, 27], le facteur de transcription TFIIH, et même l’ARN polymérase II (ARNpol II) [24]. La protéine E2, activateur transcriptionnel des gènes viraux, peut elle aussi interagir avec des facteurs d’épissage de la famille des protéines SR ou avec des protéines associées aux snRNP [28]. CAP : capping de l’ARNm ; CTD : domaine carboxyterminal de l’ARN polymérase II.
Réciproquement, l’épissage peut modifier la transcription : la présence d’un site donneur 5’ d’épissage proche du promoteur, de même que l’élimination d’introns d’un minigène, jouent clairement un rôle dans la régulation de la transcription [23]. La présence de certains facteurs d’épissage sur l’ARNm en cours de production peut par ailleurs stimuler ou inhiber l’élongation (Figure 3). Le facteur U1 snRNP en est un exemple : capable d’interagir avec le facteur d’élongation TAT-SF1 ou le facteur général de transcription TFIIH, U1 snRNP peut stimuler à la fois l’épissage et la transcription [20, 24].
Conclusions
Au vu de ces récentes observations démontrant le couplage de la transcription et de l’épissage, il est possible que l’altération de l’un des deux événements se répercute sur l’autre. De manière tout aussi concevable, le mécanisme de régulation de la transcription pourrait gouverner des événements d’épissage spécifiques d’un tissu, d’un stade de développement ou d’un état anormal de la cellule. C’est ainsi qu’il serait intéressant, par exemple, de savoir si l’oncogène Spi-1/PU.1 inhibe directement un événement d’épissage spécifique de la différenciation érythroïde terminale, comme certaines données le suggèrent [19], ou indirectement par blocage de facteurs clés pour la différenciation érythroïde terminale, tels GATA-1, NF-E2 ou EKLF. En somme, il serait déterminant de pouvoir répondre à la question suivante : Spi-1/PU.1 est-il un facteur de transcription et un facteur d’épissage, ou est-il un facteur d’épissage parce qu’il est un facteur de transcription ?
En tout état de cause, la conception de nouvelles molécules thérapeutiques ou la proposition de nouveaux traitements des cancers devrait dorénavant prendre en compte le fait que des altérations de l’expression d’un oncogène peuvent engendrer des anomalies d’épissage sur des prémessagers de régulateurs cellulaires clés, et contribuer ainsi à augmenter le potentiel tumorigène de l’oncogène.
Appendices
Remerciements
Les auteurs remercient la Ligue contre le Cancer, Comité de la Loire et Comité de la Drôme, pour leur soutien.
Références
- 1. Gisselbrecht S. Oncogenes and leukemia : history and perspectives. Med Sci (Paris) 2003 ; 19 : 201-10.
- 2. Boyd KE, Farnham PJ. Identification of target genes of oncogenic transcription factors. Proc Soc Exp Biol Med 1999 ; 222 : 9-28.
- 3. Look AT. Oncogenic transcription factors in the human acute leukemias. Science 1997 ; 278 : 1059-64.
- 4. Dittmer J, Nordheim A. Ets transcription factors and human disease. Biochim Biophys Acta 1998 ; 1377 : F1-11.
- 5. Galiana-Arnoux D, Lejeune F, Gesnel MC, et al. The CD44 alternative v9 exon contains a splicing enhancer responsive to the SR proteins 9G8, ASF/SF2, and SRp20. J Biol Chem 2003 ; 278 : 32943-53.
- 6. Caballero OL, de Souza SJ, Brentani RR, Simpson AJ. Alternative spliced transcripts as cancer markers. Dis Markers 2001 ; 17 : 67-75.
- 7. Knoop LL, Baker SJ. The splicing factor U1C represses EWS/FLI-mediated transactivation. J Biol Chem 2000 ; 275 : 24865-71.
- 8. Poleev A, Hartmann A, Stamm S. A trans-acting factor, isolated by the three-hybrid system, that influences alternative splicing of the amyloid precursor protein minigene. Eur J Biochem 2000 ; 267 : 4002-10.
- 9. Yang L, Embree LJ, Hickstein DD. TLS-ERG leukemia fusion protein inhibits RNA splicing mediated by serine-arginine proteins. Mol Cell Biol 2000 ; 20 : 3345-54.
- 10. Knoop LL, Baker SJ. EWS/FLI alters 5’-splice site selection. J Biol Chem 2001 ; 276 : 22317-22.
- 11. Yang L, Chansky HA, Hickstein DD. EWS.Fli-1 fusion protein interacts with hyperphosphorylated RNA polymerase II and interferes with serine-arginine protein-mediated RNA splicing. J Biol Chem 2000 ; 275 : 37612-8.
- 12. Chansky HA, Hu M, Hickstein DD, Yang L. Oncogenic TLS/ERG and EWS/Fli-1 fusion proteins inhibit RNA splicing mediated by YB-1 protein. Cancer Res 2001 ; 61 : 3586-90.
- 13. Moreau-Gachelin F, Wendling F, Molina T, et al. Spi-1/PU.1 transgenic mice develop multistep erythroleukemias. Mol Cell Biol 1996 ; 16 : 2453-63.
- 14. Starck J, Doubeikovski A, Sarrazin S, et al. Spi-1/PU.1 is a positive regulator of the Fli-1 gene involved in inhibition of erythroid differentiation in friend erythroleukemic cell lines. Mol Cell Biol 1999 ; 19 : 121-35.
- 15. Cantor AB, Orkin SH. Transcriptional regulation of erythropoiesis : an affair involving multiple partners. Oncogene 2002 ; 21 : 3368-76.
- 16. Hallier M, Tavitian A, Moreau-Gachelin F. The transcription factor Spi-1/PU.1 binds RNA and interferes with the RNA-binding protein p54nrb. J Biol Chem 1996 ; 271 : 11177-81.
- 17. Hallier M, Lerga A, Barnache S, et al. The transcription factor Spi-1/PU.1 interacts with the potential splicing factor TLS. J Biol Chem 1998 ; 273 : 4838-42.
- 18. Deguillien M, Huang SC, Morinière M, et al. Multiple cis elements regulate an alternative splicing event at 4.1R pre-mRNA during erythroid differentiation. Blood 2001 ; 98 : 3809-16.
- 19. Théoleyre O, Deguillien M, Morinière M, et al. Spi-1/PU.1 but not Fli-1 inhibits erythroid-specific alternative splicing of 4.1R pre-mRNA in murine erythroleukemia cells. Oncogene 2004 ; 23 : 920-7.
- 20. Orphanides G, Reinberg D. A unified theory of gene expression. Cell 2002 ; 108 : 439-51.
- 21. Nogues G, Kadener S, Cramer P, et al. Transcriptional activators differ in their abilities to control alternative splicing. J Biol Chem 2002 ; 277 : 43110-4.
- 22. Kadener S, Fededa JP, Rosbash M, Kornblihtt AR. Regulation of alternative splicing by a transcriptional enhancer through RNA pol II elongation. Proc Natl Acad Sci USA 2002 ; 99 : 8185-90.
- 23. Furger A, O’Sullivan JM, Binnie A, et al. Promoter proximal splice sites enhance transcription. Genes Dev 2002 ; 16 : 2792-9.
- 24. Kwek KY, Murphy S, Furger A, et al. U1 snRNA associates with TFIIH and regulates transcriptional initiation. Nat Struct Biol 2002 ; 9 : 800-5.
- 25. Ge H, Si Y, Wolffe AP. A novel transcriptional coactivator, p52, functionally interacts with the essential splicing factor ASF/SF2. Mol Cell 1998 ; 2 : 751-9.
- 26. Fong YW, Zhou Q. Stimulatory effect of splicing factors on transcriptional elongation. Nature 2001 ; 414 : 929-33.
- 27. Tian H. RNA ligands generated against complex nuclear targets indicate a role for U1 snRNP in co-ordinating transcription and RNA splicing. FEBS Lett 2001 ; 509 : 282-6.
- 28. Lai MC, Teh BH, Tarn WY. A human papillomavirus E2 transcriptional activator. The interactions with cellular splicing factors and potential function in pre-mRNA processing. J Biol Chem 1999 ; 274 : 11832-41.
List of figures
Figure 1
Remaniements génomiques conduisant à un dérèglement de l’expression des oncogènes.
A. Mutagenèse par insertion rétrovirale dans des gènes hôtes. Les rectangles bleus sont les séquences codant pour la protéine finale. L’insertion du provirus aboutit à l’activation de l’expression du gène par augmentation de la quantité d’ARNm disponible pour la traduction, via une activation transcriptionnelle (a) ou une stabilisation du lot d’ARNm à partir d’un promoteur non altéré (b). B. Perturbation du contexte transcriptionnel par translocation chromosomique. Le nouveau contexte chromosomique conduit à l’activation de l’expression du gène, comme pour une insertion rétrovirale (a), ou la translocation conduit à l’apparition d’un gène de fusion entre les exons de deux gènes (b).
Figure 2
Reconnaissance de l’exon 16 alternatif du prémessager 4.1R au cours de la différenciation érythroïde.
L’exon 16, exclu du messager mature dans les précurseurs, n’est retenu qu’en fin de différenciation érythroïde [18]. Les exons 14 et 15 sont exclus à tous les stades de développement érythroïde et dans la grande majorité des types cellulaires. L’analyse par Western blot utilisant un anticorps anti-Spi-1/PU.1 révèle une expression élevée de la protéine Spi-1/PU.1 dans les cellules érythroleucémiques murines (MEL) non induites (ni), cette expression étant fortement inhibée après induction des cellules (i) vers la différenciation érythroïde terminale (la révélation par un anticorps anti-actine sert de contrôle quantitatif des protéines déposées) [19].
Figure 3
Transcription et épissage, deux événements couplés pour une meilleure régulation de l’expression des gènes.
Outre la protéine oncogénique Spi-1/PU.1, plusieurs exemples de protéines impliquées dans les deux processus, transcription et épissage, sont déjà connues : P52, un activateur transcriptionnel, est capable d’interagir avec le facteur d’épissage ASF/SF2 [25]. U1 snRNP, facteur d’épissage essentiel du splicéosome, peut se lier à plusieurs protéines impliquées dans la transcription : le facteur d’élongation TAT-SF1 [26, 27], le facteur de transcription TFIIH, et même l’ARN polymérase II (ARNpol II) [24]. La protéine E2, activateur transcriptionnel des gènes viraux, peut elle aussi interagir avec des facteurs d’épissage de la famille des protéines SR ou avec des protéines associées aux snRNP [28]. CAP : capping de l’ARNm ; CTD : domaine carboxyterminal de l’ARN polymérase II.