Abstracts
Résumé
Les ostéoclastes sont des acteurs essentiels du remodelage osseux, et des anomalies de leur différenciation ou de leur activité conduisent à l’apparition de maladies osseuses, dont des défauts de résorption osseuse qui se traduisent par l’apparition d’une ostéopétrose. Différents modèles murins développant une ostéopétrose secondaire à l’apparition de mutations spontanées ou à l’invalidation de gènes ont permis de décrypter, au moins en partie, les mécanismes impliqués dans la différenciation et l’activité des ostéoclastes. Chez l’être humain, en revanche, seules des anomalies d’activité de l’ostéoclaste ont été décrites. Trois modèles murins, les souris oc/oc, gl/gl et Clcn7-/-, présentent un phénotype proche de celui de patients atteints d’ostéopétrose maligne infantile, la forme d’ostéopétrose la plus sévère chez l’homme. Des mutations dans les gènes TCIRG1, GL et CLCN7 ont donc été recherchées chez des patients ostéopétrotiques, et retrouvées majoritairement dans l’ostéopétrose maligne infantile et dans l’ostéopétrose de type II. Une telle correspondance phénotypique et génétique fait de ces trois mutants de souris des modèles particulièrement adaptés à l’étude de l’ostéopétrose humaine.
Summary
The osteoclast is the main effector of bone resorption. Failure in osteoclast differentiation or function leads to osteopetrosis, a bone disease characterized by an impaired bone resorption. Analysis of mouse models developing osteopetrosis as a consequence of naturally occuring mutations or gene knockouts allowed to establish the osteoclast differentiation pathway. Among these models, the oc/oc, the gl/gl and the Clcn7–/– mice present a phenotype similar to the one displayed by patients with infantile malignant osteopetrosis, the most severe form of osteopetrosis in human. Analysis of these models led to the identification of different mutations in the corresponding human genes TCIRG1, GL and CLCN7, in osteopetrotic patients. Mutations in the TCIRG1 gene seem the most frequent cause of malignant osteopetrosis and mutations in the CLCN7 gene seem the most frequent cause of type II osteopetrosis. Therefore, these three mouse models appear to be particularly well suited for the study of the osteoclast function in order to provide new insights in the therapy of osteopetrosis.
Article body
L’os est un tissu en perpétuel renouvellement, et l’homéostasie osseuse dépend de l’équilibre d’action de deux types cellulaires: les ostéoblastes, qui synthétisent la matrice osseuse, et les ostéoclastes, qui la résorbent. L’équilibre entre synthèse et résorption est très finement réglé, et tout déséquilibre conduit à des maladies osseuses. Chez l’homme, le terme d’ostéopétrose recouvre des maladies distinctes et hétérogènes, qui sont toutes des maladies rares associées à un défaut de résorption osseuse, avec augmentation ou non de l’activité des ostéoblastes (ostéosclérose).
L’ostéoclaste est, quant à lui, une cellule multinucléée, formée par la fusion de cellules dérivées de précurseurs de la lignée myélomonocytaire communs aux macrophages, aux cellules dendritiques et aux ostéoclastes. Sous l’influence de facteurs sécrétés par les ostéoblastes ou les cellules stromales du microenvironnement osseux, ces précurseurs se différencient en ostéoclastes (pour revue, voir [1]) ((→) m/s 2001, n° 12, p. 1260) (Figure 1).
Figure 1
Étapes de la différenciation ostéoclastique.

Les ostéoclastes sont issus de précurseurs de la lignée myélomonocytaire. Deux facteurs sont essentiels à l’ostéoclastogenèse, M-CSF (macrophage-colony stimulating factor) et RANK-L (receptor activator of nuclear factor κB ligand). Ces facteurs sont synthétisés par les cellules du microenvironnement osseux et se fixent sur leurs récepteurs respectifs c-FMS et RANK, à la surface des préostéoclastes. Ceux-ci fusionnent pour donner des ostéoclastes multinucléés qui se polarisent au contact de l’os en formant une membrane plissée, siège de la résorption osseuse. L’ostéoprotégérine (OPG) agit comme régulateur de l’ostéoclastogenèse en se fixant à RANK-L et en bloquant son effet sur RANK.
Modèles d’ostéopétrose chez la souris
Selon leur niveau d’intervention dans le processus de différenciation ostéoclastique, les gènes mutés dans les modèles murins d’ostéopétrose entraînent soit une absence (ou une diminution) de la formation des ostéoclastes, soit un défaut de fonction des ostéoclastes. Certains de ces gènes ont été retrouvés mutés dans différentes maladies osseuses humaines, qu’il s’agisse d’ostéopétrose ou d’autres affections.
Ostéopétrose liée à un défaut de différenciation des ostéoclastes
Les modèles murins de mutations spontanées ou d’invalidation de gènes, ainsi que les phénotypes qui en résultent, sont présentés dans le Tableau I.
Trois modèles animaux présentant un arrêt ou une diminution de la différenciation des progéniteurs myéloïdes en précurseurs myélomonocytaires ont permis de démontrer que le facteur de transcription PU.1, exprimé par les cellules hématopoïétiques [2], ainsi que le couple M-CSF (macrophage-colony stimulating factor)/c-FMS(récepteur du M-CSF, présent à la surface des précurseurs myélomonocytaires) [3, 4], sont essentiels à la première étape de l’ostéoclastogenèse. À l’issue de cette première phase de différenciation, les précurseurs myélomonocytaires peuvent s’engager dans différentes voies pour donner des macrophages, des ostéoclastes ou des cellules dendritiques. Cet engagement est contrôlé, au moins en partie, par c-Fos, facteur de transcription appartenant au complexe AP1, qui favoriserait la voie ostéoclastique par rapport aux voies macrophagique et dendritique; en effet, les souris c-Fos–/– ont plus de macrophages que les souris témoins [5], et il a été démontré, in vitro, que c-Fos active la différenciation des ostéoclastes et inhibe celle des cellules dendritiques [6].
La fin des années 1990 a vu se dessiner le rôle primordial, dans l’ostéoclastogenèse, du ligand de RANK (RANK-L), appartenant à la superfamille du TNF (tumor necrosis factor), de son récepteur RANK (receptor activator of nuclear factor κB) et de l’ostéoprotégérine (OPG) (Figure 2). Alors que RANK-L, synthétisé par les cellules stromales, est un activateur de la différenciation par l’intermédiaire de sa fixation au récepteur RANK présent à la surface des précurseurs ostéoclastiques, l’ostéoprotégérine, facteur soluble synthétisé par les cellules du microenvironnement osseux, inhibe les interactions entre RANK-L et RANK et module donc la différenciation des précurseurs ostéoclastiques [7]. La mutation des gènes Rank-L ou Rank se traduit par un déséquilibre du métabolisme osseux et par des atteintes du système immunitaire [8, 9].
Figure 2
Voies de signalisation impliquées dans la différenciation et l’activité des ostéoclastes.
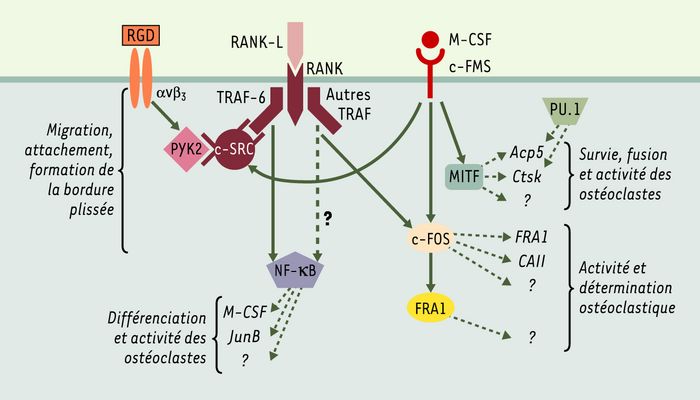
Les deux voies essentielles sont celles de RANK-L (receptor activator of nuclear factor κB ligand) et de M-CSF (macrophage-colony stimulating factor). Ces facteurs se lient sur leurs récepteurs membranaires à activité tyrosine-kinase, RANK et c-FMS, déclenchant des cascades de signalisation aboutissant à l’activation de facteurs de transcription contrôlant l’expression de gènes nécessaires à la différenciation et à l’activité de l’ostéoclaste. αvβ3: récepteur de la vitronectine; Acp5: gène codant pour la phosphatase acide résistante au tartrate; Ctsk: gène codant pour la cathepsine K; FRA1: fos-related antigen; CAII: anhydrase carbonique de type II; MITF: microphthalmia-associated transcription factor; PU.1: (Spi-1) spleen focus forming virus proviral integration oncogene 1; RGD: motifs Arg-Gly-Asp communs aux ligands des intégrines; TRAF: tumor necrosis factor receptor-associated factor; PYK2: prolin-rich tyrosine kinase.
La liaison de RANK-L sur RANK déclenche une cascade de signalisation impliquant en particulier l’activation de NF-κB (Figure 2). Ce facteur de transcription est un dimère constitué de plusieurs combinaisons de protéines très proches structurellement, p50 (NF-κB1), p52 (NF-κB2), p65 (RelA), c-Rel (Rel) et RelB. L’invalidation de chacun des gènes correspondants conduit à des anomalies de fonctionnement du système immunitaire chez la souris [10, 11]. Cependant, et malgré l’importance de la voie de signalisation de NF-κB dans l’ostéoclastogenèse, aucune atteinte du métabolisme osseux n’a été rapportée chez ces animaux. En revanche, l’observation d’une ostéopétrose chez les doubles mutants p50-/- p52-/- suggère que les deux protéines p50 et p52, dont les séquences primaires sont très similaires, ont vraisemblablement une action redondante [12, 13].
Ostéopétrose liée à un défaut de la fonction des ostéoclastes
Dans cette seconde catégorie de modèles d’ostéopétrose, les ostéoclastes sont présents, mais inactifs en raison d’un défaut affectant des protéines impliquées dans différentes fonctions: adhérence et mobilité des ostéoclastes, résorption de la matrice osseuse organique et minérale.
Les TRAF (TNF receptor-associated factors) jouent un rôle essentiel dans l’activité des ostéoclastes (Figure 2 et Tableau I). Le recrutement de TRAF-6 après la liaison de RANK-L sur son récepteur aboutit ainsi à l’activation de NF-κB, ainsi que de c-Src, une protéine à activité tyrosine-kinase également impliquée dans les cascades de signalisation du M-CSF. Cette protéine intervient également dans les voies de signalisation des intégrines, qui contrôlent l’attachement de l’ostéoclaste à l’os. La principale intégrine de l’ostéoclaste est le récepteur de la vitronectine, αvβ3. Son activation par liaison aux protéines matricielles entraîne l’activation de PYK2 (prolin-rich tyrosine kinase), une kinase qui phosphoryle c-Src. Cette association PYK2/c-Src intervient dans la régulation de l’adhérence et de la migration des ostéoclastes (pour revue, voir [14]) ((→) m/s 2001, n°12, p. 1260). Les souris chez lesquelles ces gènes ont été invalidés présentent un défaut d’attachement de l’ostéoclate à l’os [15-17]. De son côté, la protéine MITF (microphtalmia transcription factor), activée par une phosphorylation induite par le M-CSF (Figure 2), contrôle l’expression de gènes impliqués dans la survie, la fusion et l’activité des ostéoclastes [18, 19]. Le phénotype des animaux portant une mutation du gène Mitf est proche de celui des souris gl/gl qui développent à la fois une anomalie de pigmentation et une ostéopétrose sévère. Les ostéoclastes de ces souris présentent des altérations du cytosquelette et de la formation de la bordure plissée. Le gène gl muté chez ces souris code pour une protéine cytoplasmique dont le rôle n’est pas encore défini [20].
Tableau I
Modèles d’ostéopétrose décrits chez la souris et leur correspondance, quand elle existe, chez l’homme.

L’invalidation ou la mutation spontanée des gènes décrits dans ce tableau entraîne le développement d’une ostéopétrose qui peut être soit sévère, avec une absence de résorption osseuse se traduisant par une augmentation généralisée de la densité osseuse, une absence d’éruption dentaire et une létalité précoce, soit modérée, avec une diminution de la résorption osseuse se manifestant par une augmentation modérée de la densité osseuse, sans létalité.
Les voies de signalisation de RANK et du M-CSF contrôlent donc non seulement des étapes essentielles de la différenciation, mais également de l’adhérence, de la migration et de l’activité de l’ostéoclaste. Les facteurs de transcription activés par les signaux des voies de RANK-L et M-CSF contrôlent, entre autres, l’expression de gènes codant pour les enzymes de dégradation de la matrice organi-que osseuse. Parmi ces enzymes, la phosphatase acide résistante au tartrate (TRAP) participe à la déphosphorylation des protéines matricielles de l’os, et son expression est réglée par PU.1 et MITF. La cathepsine K, dont l’expression est également contrôlée par MITF, est une cystéine-protéinase participant à la dégradation de la matrice osseuse organique. L’invalidation des gènes codant pour ces deux enzymes est associée à une ostéopétrose de type modéré [21, 22].
Au contact de la surface osseuse, l’ostéoclaste se polarise et forme une membrane plissée délimitant la lacune de résorption. L’acidification de la lacune de résorption permet la dissolution de la matrice minérale et une activité optimale des enzymes protéolytiques participant à la dégradation de la matrice osseuse organique. Elle fait intervenir la pompe H+-ATPase vacuolaire (V-ATPase) et le canal chlore ClC7 (Figure3) (pour revue, voir [23]) ((→) m/s 2001, n°12, p. 1260). Ce système d’acidification est altéré dans deux modèles d’ostéopétrose, la souris ostéosclérotique oc/oc [24] et la souris Clcn7-/- [25]. Nous avons montré que les souris oc/oc portent une mutation spontanée du gène Tcirg1 (T cell immune regulator 1), également appelé Atp6i, codant pour l’isoforme α3 de la sous-unité de 116 kDa de la V-ATPase [26]. L’invalidation de ce gène reproduit le phénotype des souris oc/oc [27]. Chez la levure, deux protéines homologues de la sous-unité α, Vph1p et Stv1p, contrôlent le ciblage de la V-ATPase vers différents compartiments cytoplasmiques [28]. La perte d’association entre la V-ATPase et le cytosquelette chez la souris oc/oc suggère que la sous-unité α3 participe à la relocalisation de la V-ATPase de la vacuole vers la membrane plasmique [29].
Figure 3
Mécanismes d’acidification de la lacune de résorption.

Les protons produits par l’anhydrase carbonique de type II (CAII) passent dans les compartiments intracytoplasmiques grâce à la pompe proton-ATPase vacuolaire (V-ATPase). Cette V-ATPase est associée au canal chlore ClC7 qui permet par passage d’ions Cl–, de compenser les différences de potentiel membranaire. Ces ions Cl– entrent dans la cellule grâce à l’échangeur HCO3–/ Cl–. Lorsque l’ostéoclaste se polarise au contact de l’os, V-ATPase et ClC7, présents sur la membrane des compartiments intracytoplasmiques, sont relocalisés au niveau de la membrane plissée par fusion exocytosique, grâce à des interactions avec le cytosquelette, permettant ainsi l’acidification de la lacune de résorption.
Gènes impliqués dans l’ostéopétrose humaine
Formes d’ostéopétrose humaine
Chez l’homme, deux formes principales d’ostéopétrose sont distinguées. L’ostéopétrose maligne infantile est une maladie à transmission autosomique récessive, dont l’incidence est de 1/200000, et qui est caractérisée par des symptômes sévères liés à des anomalies du remodelage osseux (retard de croissance, malformations des os des membres et du crâne, retard à l’éruption dentaire), de l’hématopoïèse (hématopoïèse extramédullaire avec hépatosplénomégalie) et neurosensorielles (cécité et troubles du développement psychomoteur pouvant être dus à une compression osseuse ou à la maladie elle-même). En l’absence de traitement, la survie des patients est estimée à 30% à l’âge de 6 ans, avec une qualité de vie extrêmement médiocre. Infections, hémorragies et anémie rendent compte du décès pendant la première décennie. L’autre forme d’ostéopétrose, à transmission autosomique dominante, recouvre différentes maladies partageant une sémiologie osseuse commune [30], parmi lesquelles l’ostéopétrose de type II (maladie d’Albers-Schönberg) est la plus fréquente[1] (environ 1/100 000). Cette maladie reste bénigne, et de nombreux patients sont asymptomatiques: ses caractéristiques majeures sont la survenue fréquente de fractures, une ostéomyélite mandibulaire et une ostéoarthrite de la hanche.
Bases génétiques de l’ostéopétrose humaine
Les patients atteints d’ostéopétrose maligne infantile ont un phénotype équivalent à celui décrit chez la souris oc/oc(Figure 4), avec comme caractéristiques principales une densité osseuse élevée et une réduction des cavités médullaires. Des mutations du gène TCIRG1 ont donc été recherchées chez ces patients, et détectées dans environ 50 % des cas étudiés [31-34]. Si la majorité de ces mutations touche des sites donneurs ou accepteurs d’épissage, des insertions, des délétions ou des mutations non-sens ont également été observées. Des mutations ponctuelles entraînant des modifications d’acides aminés conservés dans les séquences des homologues de TCIRG1 de plusieurs espèces ont également été décrites. Ces mutations entraînent probablement la production de protéines non fonctionnelles, ou d’ARNm instables ou ne pouvant être traduits correctement [33]. Cependant, toutes ces mutations ne sont pas présentes à l’état homozygote: certains patients sont des hétérozygotes composites et, dans certains cas, des mutations ne sont détectées que sur un seul allèle du gène. Dans tous les cas étudiés, les régions promotrices et les introns du gène n’ont pas été analysés de façon extensive. L’ostéopétrose maligne infantile ayant une transmission de type autosomique récessif, la présence de mutations dans les régions régulatrices ou introniques du gène TCIRG1 ne peut être exclue.
Figure 4
Radiographies du squelette d’une souris ostéosclérotique oc/oc et d’un patient atteint d’ostéopétrose maligne infantile.
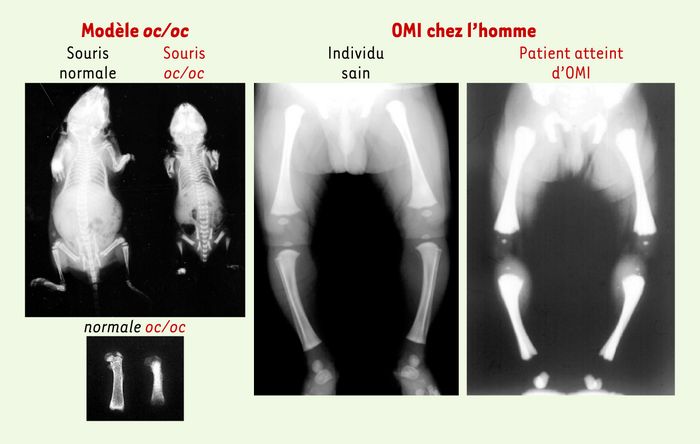
L’analyse comparative par radiographie du squelette d’une souris oc/oc et d’une souris normale âgées de 15 jours met en évidence chez la souris oc/oc un retard de croissance, une augmentation de la densité osseuse et une absence de cavité médullaire dans les os longs. Ces anomalies des os longs sont retrouvées sur la radiographie du squelette d’un enfant atteint d’ostéopétrose maligne infantile, en comparaison avec un enfant sain du même âge (2 semaines).
Des mutations du gène CLCN7 ont également été recherchées et détectées dans neuf familles de patients atteints d’ostéopétrose maligne infantile [25, 35, 36]. Elles consistent en insertions, délétions ou mutations non-sens et faux-sens présentes à l’état homozygote ou hétérozygote composite. Des mutations du gène CLCN7 ont également été décrites dans des familles de patients atteints d’ostéopétrose de type II [35, 36]. Ces mutations sont des délétions d’un ou deux nucléotides, ou des mutations faux-sens touchant, là encore, des acides aminés conservés chez tous les membres de la famille des protéines CLC. Cependant, les raisons pour lesquelles des patients porteurs de mutations du gène CLCN7 peuvent avoir un phénotype très sévère (ostéopétrose maligne infantile) ou plutôt bénin (type II), ainsi que des modes de transmission différents (récessif ou dominant), restent à élucider.
Récemment, une étude portant sur 19patients ne présentant pas de mutation dans les gènes TCIRG1 et CLCN7 a montré la présence d’une mutation du gène GL, sur un site donneur d’épissage, chez un seul de ces patients [20]. Les mutations du gène GL restent donc minoritaires dans l’ostéopétrose maligne infantile.
Les mutations des gènes TCIRG1 et CLCN7 étant respectivement les causes les plus fréquentes d’ostéopétrose maligne infantile et de type II, la recherche systématique de mutations dans ces gènes est donc nécessaire en cas de diagnostic d’ostéopétrose. Cependant, un certain nombre de patients ne présentent pas de mutation dans la séquence codante de ces gènes: ces données suggèrent la présence de mutations dans des régions régulatrices de ces gènes, ou l’intervention d’autres gènes encore non identifiés.
Conclusions
L’hétérogénéité phénotypique de l’ostéopétrose, rencontrée chez l’homme comme chez la souris, est probablement liée au grand nombre de gènes potentiellement impliqués dans le processus de différenciation des ostéoclastes, et au niveau où ces gènes interviennent. La biologie de l’ostéoclaste a donc largement profité de l’analyse des mutants ostéopétrotiques.
Chez la souris, les défauts de la différenciation ostéoclastique bloquent également d’autres voies de différenciation du système hématopoïétique. Chez l’homme, en revanche, seules des anomalies d’activité de l’ostéoclaste ont été décrites: les défauts de différenciation du système hématopoïétique sont donc probablement létaux à un stade précoce du développement humain.
Les souris oc/oc, gl/gl et Clcn7–/– constituent des modèles prometteurs, non seulement car ils reproduisent le phénotype des patients atteints d’ostéopétrose maligne infantile, mais aussi parce que les bases génétiques de cette ostéopétrose sont les mêmes que chez les patients. L’étude de ces modèles devrait donc permettre le développement de nouveaux agents thérapeutiques de l’ostéopétrose, mais également des maladies osseuses en général.
L’ostéopétrose maligne infantile est spontanément mortelle lorsqu’elle n’est pas traitée. Le seul traitement ayant entraîné une rémission réelle et à long terme est l’allogreffe de moelle osseuse en condition allogénique, mais son efficacité reste faible [37]. Toutefois, les progrès réalisés dans le cas de maladies touchant les cellules du système hématopoïétique [38], dont est issu l’ostéoclaste, suggèrent que la thérapie cellulaire appliquée à l’ostéoclaste pourrait être envisagée dans le cas de l’ostéopétrose maligne infantile.
Appendices
Note
-
[1]
L’ostéopétrose liée à un déficit en anhydrase carbonique de type II présente quant à elle un phénotype intermédiaire, avec une transmission autosomique récessive.Cependant, l’invalidation, chez la souris, du gène correspondant ne conduisant pas à un phénotype d’ostéopétrose, nous ne traiterons pas de cette maladie dans le cadre de cet article.
Références
- 1. Teitelbaum SL. Bone resorption by osteoclasts. Science 2000; 289: 1504-8.
- 2. Tondravi MM, McKercher SR, Anderson K, et al. Osteopetrosis in mice lacking haematopoietic transcription factor PU.1. Nature 1997; 386:81-4.
- 3. Yoshida H, Hayashi S, Kunisada T, et al. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature 1990; 345: 442-4.
- 4. Dai XM, Ryan GR, Hapel AJ, et al. Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood 2002; 99: 111-20.
- 5. Grigoriadis AE, Wang ZQ, Cecchini MG, et al. c-Fos: a key regulator of osteoclast-macrophage lineage determination and bone remodeling. Science 1994; 266: 443-8.
- 6. Miyamoto T, Ohneda O, Arai F, et al. Bifurcation of osteoclasts and dendritic cells from common progenitors. Blood 2001; 98: 2544-54.
- 7. Simonet WS, Lacey DL, Dunstan CR, et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell 1997; 89: 309-19.
- 8. Kong YY, Yoshida H, Sarosi I, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 1999; 397: 315-23.
- 9. Dougall WC, Glaccum M, Charrier K, et al. RANK is essential for osteoclast and lymph node development. Genes Dev 1999; 13: 2412-24.
- 10. Snapper CM, Zelazowski P, Rosas FR, et al. B cells from p50/NF-kappa B knockout mice have selective defects in proliferation, differentiation, germ-line CH transcription, and Ig class switching. J Immunol 1996; 156: 183-91.
- 11. Sha WC, Liou HC, Tuomanen EI, et al. Targeted disruption of the p50 subunit of NF-kappa B leads to multifocal defects in immune responses. Cell 1995; 80: 321-30.
- 12. Franzoso G, Carlson L, Xing L, et al. Requirement for NF-kappaB in osteoclast and B-cell development. Genes Dev 1997; 11: 3482-96.
- 13. Iotsova V, Caamano J, Loy J, et al. Osteopetrosis in mice lacking NF-kappaB1 and NF-kappaB2. Nat Med 1997; 3: 1285-9.
- 14. Duong LT, Rodan GA. Integrin-mediated signaling in the regulation of osteoclast adhesion and activation. Front Biosci 1998; 3: d757-68.
- 15. Lomaga MA, Yeh WC, Sarosi I, et al. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev 1999; 13: 1015-24.
- 16. Soriano P, Montgomery C, Geske R, et al. Targeted disruption of the c-src proto-oncogene leads to osteopetrosis in mice. Cell 1991; 64: 693-702.
- 17. McHugh KP, Hodivala-Dilke K, Zheng MH, et al. Mice lacking beta3 integrins are osteosclerotic because of dysfunctional osteoclasts. J Clin Invest 2000; 105 : 433-40.
- 18. Luchin A, Suchting S, Merson T, et al. Genetic and physical interactions between microphthalmia transcription factor and PU.1 are necessary for osteoclast gene expression and differentiation. J Biol Chem 2001; 276: 36703-10.
- 19. Hodgkinson CA, Moore KJ, Nakayama A, et al. Mutations at the mouse microphthalmia locus are associated with defects in a gene encoding a novel basic-helix-loop-helix-zipper protein. Cell 1993; 74: 395-404.
- 20. Chalhoub N, Benachenhou N, Rajapurohitam V, et al. Grey-lethal mutation induces severe malignant autosomal recessive osteopetrosis in mouse and human. Nat Med 2003; 9: 399-406.
- 21. Hayman AR, Jones SJ, Boyde A, et al. Mice lacking tartrate-resistant acid phosphatase (Acp 5) have disrupted endochondral ossification and mild osteopetrosis. Development 1996; 122: 3151-62.
- 22. Gowen M, Lazner F, Dodds R, et al. Cathepsin K knockout mice develop osteopetrosis due to a deficit in matrix degradation but not demineralization. J Bone Miner Res 1999; 14: 1654-63.
- 23. Nishi T, Forgac M. The vacuolar (h+)-ATPases - nature’s most versatile proton pumps. Nat Rev Mol Cell Biol 2002; 3: 94-103.
- 24. Seifert MF, Marks SC Jr. Morphological evidence of reduced bone resorption in the osteosclerotic (oc) mouse. Am J Anat 1985; 172: 141-53.
- 25. Kornak U, Kasper D, Bosl MR, et al. Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell 2001; 104: 205-15.
- 26. Scimeca JC, Franchi A, Trojani C, et al. The gene encoding the mouse homologue of the human osteoclast-specific 116-kDa V-ATPase subunit bears a deletion in osteosclerotic (oc/oc) mutants. Bone 2000; 26: 207-13.
- 27. Li YP, Chen W, Liang Y, et al. Atp6i-deficient mice exhibit severe osteopetrosis due to loss of osteoclast-mediated extracellular acidification. Nat Genet 1999; 23: 447-51.
- 28. Kawasaki-Nishi S, Bowers K, Nishi T, et al. The amino-terminal domain of the vacuolar proton-translocating ATPase a subunit controls targeting and in vivo dissociation, and the carboxyl- terminal domain affects coupling of proton transport and ATP hydrolysis. J Biol Chem 2001; 276: 47411-20.
- 29. Nakamura I, Takahashi N, Udagawa N, et al. Lack of vacuolar proton ATPase association with the cytoskeleton in osteoclasts of osteosclerotic (oc/oc) mice. FEBS Lett 1997; 401: 207-12.
- 30. Key L, Ries W. Osteopetrosis. In: Bilezikian J, Raisz L, Rodan G, eds. Principles of bone biology. New York: Academic Press, 2002: 1217-27.
- 31. Kornak U, Schulz A, Friedrich W, et al. Mutations in the a3 subunit of the vacuolar H(+)-ATPase cause infantile malignant osteopetrosis. Hum Mol Genet 2000; 9: 2059-63.
- 32. Frattini A, Orchard PJ, Sobacchi C, et al. Defects in TCIRG1 subunit of the vacuolar proton pump are responsible for a subset of human autosomal recessive osteopetrosis. Nat Genet 2000; 25: 343-6.
- 33. Sobacchi C, Frattini A, Orchard P, et al. The mutational spectrum of human malignant autosomal recessive osteopetrosis. Hum Mol Genet 2001; 10: 1767-73.
- 34. Scimeca JC, Quincey D, Parrinello H, et al. New mutations in the gene encoding the a3 subunit of the vacuolar proton pump in patients affected by infantile malignant osteopetrosis. Hum Mutat 2003; 21: 151-7.
- 35. Cleiren E, Benichou O, Van Hul E, et al. Albers-Schonberg disease (autosomal dominant osteopetrosis, type II) results from mutations in the ClCN7 chloride channel gene. Hum Mol Genet 2001; 10: 2861-7.
- 36. Frattini A, Pangrazio A, Susani L, et al. Chloride channel ClCN7 mutations are responsible for severe recessive, dominant, and intermediate osteopetrosis. J Bone Miner Res 2003; 18: 1740-7.
- 37. Gerritsen EJ, Vossen JM, Fasth A, et al. Bone marrow transplantation for autosomal recessive osteopetrosis. A report from the working party on inborn errors of the European bone marrow transplantation group. J Pediatr 1994; 125: 896-902
- 38. Cavazzana-Calvo M, Hacein-Bey S, de Saint Basile G, et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science 2000; 288: 669-72.
- 39. Hughes AE, Ralston SH, Marken J, et al. Mutations in TNFRSF11A, affecting the signal peptide of RANK, cause familial expansile osteolysis. Nat Genet 2000; 24: 45-8.
- 40. Whyte M, Obrecht S, Finnegan P, et al. Osteoprotegerin deficiency and juvenile Paget’s disease. J Exp Med 2002; 347: 175-84
- 41. Gelb BD, Shi GP, Chapman HA, et al. Pycnodysostosis, a lysosomal disease caused by cathepsin K deficiency. Science 1996; 273: 1236-8.
List of figures
Figure 1
Étapes de la différenciation ostéoclastique.
Les ostéoclastes sont issus de précurseurs de la lignée myélomonocytaire. Deux facteurs sont essentiels à l’ostéoclastogenèse, M-CSF (macrophage-colony stimulating factor) et RANK-L (receptor activator of nuclear factor κB ligand). Ces facteurs sont synthétisés par les cellules du microenvironnement osseux et se fixent sur leurs récepteurs respectifs c-FMS et RANK, à la surface des préostéoclastes. Ceux-ci fusionnent pour donner des ostéoclastes multinucléés qui se polarisent au contact de l’os en formant une membrane plissée, siège de la résorption osseuse. L’ostéoprotégérine (OPG) agit comme régulateur de l’ostéoclastogenèse en se fixant à RANK-L et en bloquant son effet sur RANK.
Figure 2
Voies de signalisation impliquées dans la différenciation et l’activité des ostéoclastes.
Les deux voies essentielles sont celles de RANK-L (receptor activator of nuclear factor κB ligand) et de M-CSF (macrophage-colony stimulating factor). Ces facteurs se lient sur leurs récepteurs membranaires à activité tyrosine-kinase, RANK et c-FMS, déclenchant des cascades de signalisation aboutissant à l’activation de facteurs de transcription contrôlant l’expression de gènes nécessaires à la différenciation et à l’activité de l’ostéoclaste. αvβ3: récepteur de la vitronectine; Acp5: gène codant pour la phosphatase acide résistante au tartrate; Ctsk: gène codant pour la cathepsine K; FRA1: fos-related antigen; CAII: anhydrase carbonique de type II; MITF: microphthalmia-associated transcription factor; PU.1: (Spi-1) spleen focus forming virus proviral integration oncogene 1; RGD: motifs Arg-Gly-Asp communs aux ligands des intégrines; TRAF: tumor necrosis factor receptor-associated factor; PYK2: prolin-rich tyrosine kinase.
Figure 3
Mécanismes d’acidification de la lacune de résorption.
Les protons produits par l’anhydrase carbonique de type II (CAII) passent dans les compartiments intracytoplasmiques grâce à la pompe proton-ATPase vacuolaire (V-ATPase). Cette V-ATPase est associée au canal chlore ClC7 qui permet par passage d’ions Cl–, de compenser les différences de potentiel membranaire. Ces ions Cl– entrent dans la cellule grâce à l’échangeur HCO3–/ Cl–. Lorsque l’ostéoclaste se polarise au contact de l’os, V-ATPase et ClC7, présents sur la membrane des compartiments intracytoplasmiques, sont relocalisés au niveau de la membrane plissée par fusion exocytosique, grâce à des interactions avec le cytosquelette, permettant ainsi l’acidification de la lacune de résorption.
Figure 4
Radiographies du squelette d’une souris ostéosclérotique oc/oc et d’un patient atteint d’ostéopétrose maligne infantile.
L’analyse comparative par radiographie du squelette d’une souris oc/oc et d’une souris normale âgées de 15 jours met en évidence chez la souris oc/oc un retard de croissance, une augmentation de la densité osseuse et une absence de cavité médullaire dans les os longs. Ces anomalies des os longs sont retrouvées sur la radiographie du squelette d’un enfant atteint d’ostéopétrose maligne infantile, en comparaison avec un enfant sain du même âge (2 semaines).
List of tables
Tableau I
Modèles d’ostéopétrose décrits chez la souris et leur correspondance, quand elle existe, chez l’homme.
L’invalidation ou la mutation spontanée des gènes décrits dans ce tableau entraîne le développement d’une ostéopétrose qui peut être soit sévère, avec une absence de résorption osseuse se traduisant par une augmentation généralisée de la densité osseuse, une absence d’éruption dentaire et une létalité précoce, soit modérée, avec une diminution de la résorption osseuse se manifestant par une augmentation modérée de la densité osseuse, sans létalité.