Article body
Le génome humain présente des polymorphismes fréquents, en moyenne tous les 1000 nucléotides. Les plus représentés de ces polymorphismes ont pour origine le remplacement d’un nucléotide par un autre (SNP pour single nucleotide polymorphism). Il en est ainsi parce qu’à chaque génération, de nouveaux variants nucléotidiques apparaissent à la suite de mutations survenues dans les gamètes parentaux. La plupart de ces mutations n’ont pas d’effet sur le phénotype et seront perpétuées ou disparaîtront de façon essentiellement aléatoire. Certaines, parce qu’elles sont défavorables aux individus qui les portent, seront inéluctablement éliminées. D’autres, enfin, parce qu’elles confèrent un avantage dans des conditions données, verront leur fréquence augmenter dans une population.
Une méthode générale permettant d’identifier des gènes qui ont été soumis dans une période récente à une pression de sélection positive vient d’être proposée [1].
Elle repose sur cinq concepts principaux.
On observe que des polymorphismes présents dans une même région du génome (de l’ordre de quelques milliers de nucléotides) ne sont pas associés au hasard: la présence sur un même chromosome des allèles correspondant à plusieurs SNP définit un nombre restreint d’haplotypes. L’observation d’un nombre d’haplotypes inférieur au nombre d’haplotypes théoriquement obtenus par combinaison aléatoire des allèles des différents polymorphismes traduit l’existence d’un « déséquilibre de liaison ».
L’importance quantitative des déséquilibres de liaison diminue avec la distance entre les polymorphismes, et avec le temps. En effet, les recombinaisons se produisant à chaque méiose font que, à chaque génération, il existe une probabilité que l’association entre des allèles particuliers présents sur un même chromosome soit modifiée.
En l’absence de sélection, un allèle ne peut atteindre une fréquence élevée par simple dérive génétique qu’après un grand nombre de générations, et cela d’autant plus que la taille de la population est grande. Les allèles fréquents sont donc le plus souvent anciens.
La fréquence d’un allèle peut subir une augmentation rapide du fait d’une forte sélection. Dans ce cas, les allèles des polymorphismes qui étaient présents sur le même chromosome, à proximité de l’allèle sélectionné, vont voir également leur fréquence augmenter rapidement: c’est l’effet « auto-stop » qui explique que des déséquilibres de liaison puissent être observés entre l’allèle soumis à sélection et des polymorphismes relativement éloignés, car le petit nombre de générations depuis l’apparition de l’allèle sélectionné n’a pas laissé le temps de « battre les cartes » par recombinaison.
La combinaison de plusieurs polymorphismes proches, définissant n haplotypes, peut être considérée comme un locus polymorphe unique présentant n allèles différents.
Pour déterminer si un gène a été récemment soumis à une pression de sélection, les auteurs proposent donc d’identifier des haplotypes appelés core haplotypes en génotypant des polymorphismes au sein d’un petit intervalle contenant le gène, et de mesurer le déséquilibre de liaison entre chacun de ces haplotypes et des marqueurs plus distants (Figure 1). Le stigmate d’une sélection récente est l’existence d’un déséquilibre de liaison entre l’haplotype sélectionné et des marqueurs distants significativement plus grand que le déséquilibre de liaison existant entre les autres core haplotypes et les mêmes marqueurs distants.
Figure 1
Localisation approximative des polymorphismes étudiés au voisinage du gène de la G6PD (glucose-6-phosphate déshydrogénase).
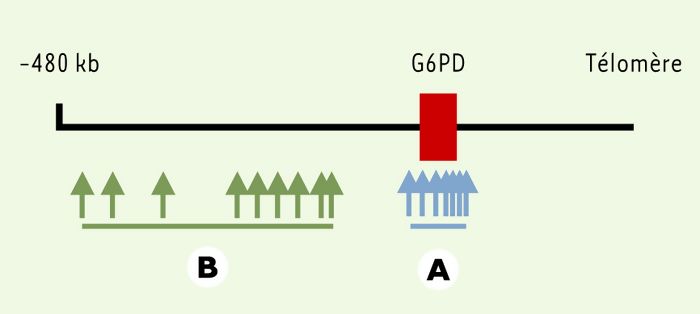
A. Polymorphismes définissant les core haplotypes du gène de la G6PD. B. Polymorphismes à distance du gène permettant de mesurer le déséquilibre de liaison avec les core haplotypes.
Un des avantages de cette méthode est de s’affranchir de la difficulté venant de l’absence d’homogénéité des recombinaisons sur le génome en prenant comme « témoins internes » les autres core haplotypes.
Pour tester la validité de cette approche, les auteurs l’ont appliquée à deux gènes dont la sélection positive était déjà établie: le gène de la G6PD, codant pour une enzyme du globule rouge, la glucose-6-phosphate déshydrogénase (Figure 1) et le gène codant pour le ligand de CD40 (ou TNFSF5, codant pour une protéine de la famille du tumor necrosis factor). De nombreuses études épidémiologiques avaient fortement suggéré que des variants fréquents de ces gènes confèrent une protection partielle contre le paludisme. Pour chacun de ces gènes, la méthode a confirmé - à partir de l’étude de plusieurs populations africaines - qu’un haplotype unique avait été soumis à une pression de sélection positive au cours de l’histoire récente de l’humanité: (les estimations proposées sont de 2500 ans pour le variant de la G6PD et de 6500 ans pour le variant du ligand de CD40. À l’inverse, aucun indice en faveur d’une sélection d’allèle n’a été trouvé pour 17 locus choisis au hasard et étudiés chez les mêmes populations.
Les perspectives de ce travail sont d’identifier à l’échelle du génome humain entier les régions qui ont été, au cours d’un passé récent, soumises à une pression de sélection. La condition est, évidemment, que les variants récemment apparus et en expansion rapide n’aient pas eu le temps d’être fixés, c’est-à-dire d’avoir supplanté les autres allèles. En cas de fixation d’un allèle récemment sélectionné, on s’attendrait à trouver dans la région soumise à sélection un « déficit » de polymorphismes au sein des populations soumises à la sélection. Il est intéressant de noter que la répartition des polymorphismes sur le génome montre l’existence d’une répartition qui n’est pas aléatoire [2]. Une fraction notable du génome présente une faible densité en polymorphismes qui pourrait s’expliquer, en partie, par une sélection récente de certaines de ces régions.
Un des intérêts d’identifier de manière systématique les gènes récemment soumis à sélection dans certaines populations pourrait être de fournir des pistes de recherche de facteurs génétiques protecteurs vis-à-vis de maladies fréquentes. Cela requiert, bien entendu, de confronter les données génétiques aux résultats d’études historiques, archéologiques et anthropologiques, aux connaissances physiopathologiques, afin de formuler des hypothèses pertinentes qui devront ensuite être testées de différentes manières.
Appendices
Références
- 1. Sabeti PC, Reich DE, Higgins JM, et al. Detecting recent positive selection in the human genome from haplotype structure. Nature 2002; 419: 832-7.
- 2. Venter JC, Adams MD, Myers EW, et al. The sequence of the human genome. Science 2001; 291: 1304-51.
List of figures
Figure 1
Localisation approximative des polymorphismes étudiés au voisinage du gène de la G6PD (glucose-6-phosphate déshydrogénase).
A. Polymorphismes définissant les core haplotypes du gène de la G6PD. B. Polymorphismes à distance du gène permettant de mesurer le déséquilibre de liaison avec les core haplotypes.