Article body
Les maladies par expansion de polyglutamine tiennent une place particulière parmi les maladies neurodégénératives. Elles constituent un groupe de neuf maladies héréditaires, comprenant la maladie de Huntington et plusieurs formes d’ataxies spinocérébelleuses, dues à une expansion de triplets CAG dans la séquence codante de neuf gènes différents [1, 2]. Le seuil pathologique apparait similaire avec des allèles mutés portant plus de ~ 30-40 triplets CAG, et la sévérité de la maladie est corrélée à la taille de l’expansion. La mutation se traduit par la synthèse d’une protéine avec une expansion d’un motif polyglutamine provoquant un dysfonctionnement et une mort neuronale. Cependant, les mécanismes expliquant la toxicité neuronale des expansions de polyglutamine sont encore mal compris. Malgré les propriétés communes de ces mutations et l’expression ubiquitaire des gènes portant l’expansion CAG, chaque maladie se distingue par une sensibilité particulière de certains types de neurones.
Les expansions de polyglutamine entraînent une toxicité par gain de fonction, faisant appel à des mécanismes communs, liés directement à l’expansion de polyglutamine. Par ailleurs, des mécanismes propres à chaque maladie, liés au contexte protéique dans lequel se situe le motif polyglutamine, expliquent la spécificité de l’atteinte neuronale dans ces maladies. Il paraît donc crucial d’identifier précisément la fonction de ces protéines afin de déterminer si elle est altérée par la mutation. À cet égard, l’étude de l’ataxie spinocérébelleuse de type 7 (SCA7) a permis d’identifier de nouvelles anomalies induites par les expansions de polyglutamine. En effet, nous avons récemment montré que l’ataxine-7 (ATXN7), la protéine mutée dans SCA7, est une nouvelle sous-unité d’un complexe transcriptionnel, TFTC/STAGA (TBP-free TAF-containing complex/SPT3/TAF9/GCN5 acetyltransferase complex) [3]. TFTC/STAGA est un co-activateur de la transcription, servant de pont entre un facteur de transcription lié à son promoteur et l’ARN polymérase II. TFTC/STAGA stimule également la transcription grâce à l’activité d’acétylation de l’histone H3 porté par la sous-unité GCN5. La fonction de l’ATXN7 était donc clairement identifiée et ce résultat a ensuite été confirmé par deux autres groupes [4, 5].
Ataxie spinocérébelleuse de type 7 et atteinte rétinienne
D’autre part, SCA7 est la seule maladie par expansion de polyglutamine dans laquelle la rétine est atteinte [6]. Plusieurs groupes ont analysé la toxicité de l’ATXN7 mutée dans la rétine de souris transgéniques et identifié des anomalies transcriptionnelles reproductibles entre les différents modèles utilisés [7-9]. Des anomalies transcriptionnelles ont également été observées dans d’autres modèles de maladies par expansion de polyglutamine, mais les mécanismes en cause et les liens avec les phénotypes observés demeurent obscurs dans la plupart des cas [10]. L’analyse de la rétinopathie dans les modèles SCA7 a montré une diminution de l’expression de gènes spécifiques des photorécepteurs. En particulier, la perte des protéines de la transduction du signal lumineux, explique la perte de l’activité des photorécepteurs des souris modèles. L’ensemble de ces observations offrait alors la possibilité d’étudier comment l’expansion de polyglutamine modifie la fonction normale de l’ATXN7 à l’intérieur du complexe TFTC/STAGA et comment ceci affecte l’expression des gènes impliqués dans l’activité des photorécepteurs.
Dérèglement du complexe transcriptionnel TFTC/STAGA et modification de la chromatine
Plusieurs études récentes suggèrent en effet que le dysfonctionnement des photorécepteurs dans différents modèles de SCA7 est lié à une fonction anormale de TFTC/STAGA entraînant des modifications de la chromatine. L’analyse de complexes TFTC/STAGA à partir de cellules en culture ou dans la levure suggère que l’incorporation de l’ATXN7 mutée induit une diminution de l’incorporation de certaines sous-unités dans ces complexes et une perte de l’activité d’acétylation des histones [4, 5]. Ces travaux montrent également que CRX (retinal cone-rod homeobox factor), un facteur de transcription spécifique des photorécepteurs, interagit avec l’ATXN7 in vitro. Leurs résultats suggèrent que TFTC/STAGA pourrait être recruté par CRX sur les promoteurs des gènes fortement exprimés dans les photorécepteurs. Chez les patients SCA7, l’ATXN7 avec une expansion polyglutamine inhiberait l’activité d’acétylation des histones de TFTC/STAGA et le recrutement de CRX sur les promoteurs des gènes spécifiques des photorécepteurs.
Cependant ces observations sont contredites par une étude récente, fondée exclusivement sur l’analyse de la rétine de souris SCA7 dans lesquelles des anomalies transcriptionnelles caractéristiques sont à l’origine du dysfonctionnement des photorécepteurs [11]. Dans ces modèles SCA7, nous avons identifié des modifications extrêmement sévères de la structure de la chromatine. Alors que les photorécepteurs normaux présentent une large région d’hétérochromatine centrale et un fin liseré d’euchromatine périphérique, on observe un élargissement considérable des noyaux des photorécepteurs avec une décondensation majeure du territoire d’hétérochromatine centrale dans la rétine des souris SCA7 (Figure 1). Ces modifications sont précoces et leur progression est directement corrélée à la répression de l’expression des gènes spécifiques des photorécepteurs.
Figure 1
Réorganisation de la chromatine et perte de l’expression des gènes spécifiques des photorécepteurs dans la rétine des souris SCA7.
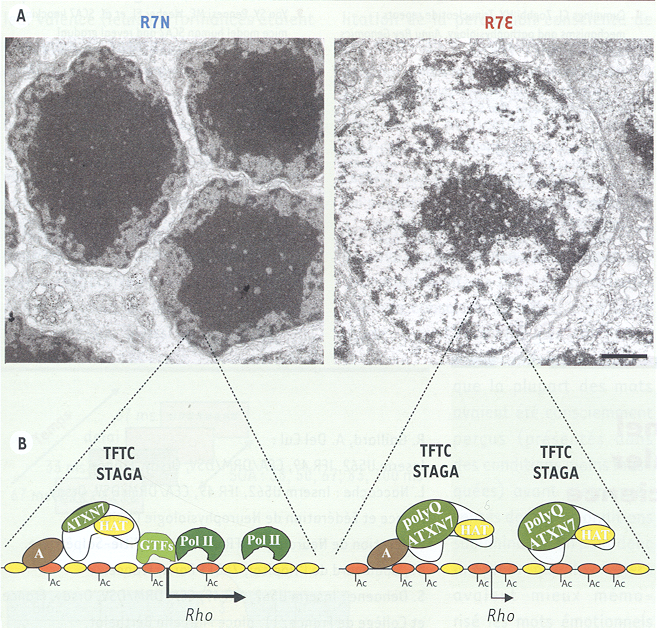
A. Images de microscopie électronique des noyaux des photorécepteurs de type bâtonnets dans les souris témoins (R7N) et SCA7 (R7E). Les bâtonnets normaux sont caractérisés par une large région d’hétérochromatine centrale et un liseré d’euchromatine périphérique. L’ATXN7 mutée provoque une augmentation très importante du volume nucléaire et une décondensation majeure de l’hétérochromatine centrale. B. Modèle des effets d’une expansion polyglutamine dans ATXN7 (bas). Dans les bâtonnets normaux, les gènes spécifiques des photorécepteurs, tel que le gène codant pour la rhodopsine, sont localisés dans la région périphérique d’hétérochromatine où une forte concentration d’activateurs (A) et de facteurs généraux de la transcription (GTF) permet d’atteindre de forts niveaux d’expression. Dans les souris SCA7, une augmentation du recrutement de TFTC/STAGA sur le promoteur provoque une hyperacétylation des histones. La décondensation de la chromatine entraîne une dilution des facteurs de transcription (A et GTF) expliquant la diminution de l’expression des gènes fortement exprimés dans les bâtonnets (adapté de [11]).
L’acétylation des histones entraînant une décondensation de la chromatine, nous avons alors analysé les complexes TFTC/STAGA dans les rétines de souris témoins et SCA7. Nous avons observé que la composition et l’activité d’acétylation des histones des complexes ayant incorporé l’ATXN7 mutée est identique à celles des complexes contenant l’ATXN7 normale. Cependant, le recrutement de TFTC/STAGA sur les promoteurs des gènes spécifiques des photorécepteurs est augmenté dans les souris SCA7 par rapport aux souris témoins et, en accord avec ce résultat, ces promoteurs sont hyperacétylés. Ce résultat est très surprenant car nous observons une diminution de l’expression des gènes dont les promoteurs sont hyperacétylés chez les souris SCA7, alors que, selon le code de modification des histones [12], l’acétylation des histones facilite la transcription. En revanche, la décondensation globale de la chromatine observée dans les photorécepteurs des souris SCA7 pourrait être causée par cette hyperacétylation, mais devrait être associée à une augmentation de l’expression d’un grand nombre de gènes. Là encore, nous avons observé l’inverse puisque l’étude du transcriptome des rétines de souris SCA7 a montré que l’expression d’une faible proportion de gènes (~ 1-2 %) est modifiée et dans le sens d’une diminution pour la plupart.
Sur la base de ces observations, nous suggérons le modèle suivant (Figure 1) : l’incorporation de l’ATXN7 mutée dans les complexes TFTC/STAGA induit un recrutement aberrant de ces complexes aux gènes spécifiques des photorécepteurs. Cela induit une hyperacétylation des histones, la décondensation de la chromatine et une augmentation de la taille du noyau des photorécepteurs entraînant une « dilution » des facteurs de transcription. Cet effet de « dilution » conduirait à une diminution de l’expression des gènes qui sont normalement spécifiquement et fortement exprimés dans la rétine. Le contrôle de leur expression serait le plus sensible à une perturbation de l’architecture particulière des noyaux des photorécepteurs. Cette hypothèse est renforcée par l’observation d’une distribution des gènes fortement exprimés dans l’euchromatine périphérique, alors que des gènes non exprimés sont localisés dans l’hétérochromatine centrale. Cette répartition est perdue dans les photorécepteurs des souris SCA7, expliquant ainsi le profil d’expression des gènes observé chez ces animaux. En conclusion, ces études montrent la nécessité de comprendre la fonction normale des protéines portant l’expansion de polyglutamine et indiquent que des modifications de la chromatine pourraient jouer un rôle majeur dans les maladies neurodégénératives.
Appendices
Références
- 1. Cummings CJ, Zoghbi HY. Trinucleotide repeats : mechanisms and pathophysiology. Annu Rev Genomics Hum Genet 2000 ; 1 : 281-328.
- 2. Lebre A, Brice A. Maladies par expansion de polyglutamine. Données moléculaires et physiopathologiques. Med Sci (Paris) 2001 ; 17 : 1149-57.
- 3. Helmlinger D, Hardy S, Sasorith S, et al. Ataxin-7 is a subunit of GCN5 histone acetyltransferase-containing complexes. Hum Mol Genet 2004 ; 13 : 1257-65.
- 4. McMahon SJ, Pray-Grant MG, Schieltz D, et al. Polyglutamine-expanded spinocerebellar ataxia-7 protein disrupts normal SAGA and SLIK histone acetyltransferase activity. Proc Natl Acad Sci USA 2005 ; 102 : 8478-82.
- 5. Palhan VB, Chen S, Peng GH, et al. Polyglutamine-expanded ataxin-7 inhibits STAGA histone acetyltransferase activity to produce retinal degeneration. Proc Natl Acad Sci USA 2005 ; 102 : 8472-7.
- 6. Michalik A, Martin JJ, Van Broeckhoven C. Spinocerebellar ataxia type 7 associated with pigmentary retinal dystrophy. Eur J Hum Genet 2004 ; 12 : 2-15.
- 7. Helmlinger D, Abou-Sleymane G, Yvert G, et al. Disease progression despite early loss of polyglutamine protein expression in SCA7 mouse model. J Neurosci 2004 ; 24 : 1881-7.
- 8. La Spada AR, Fu YH, Sopher BL, et al. Polyglutamine-expanded ataxin-7 antagonizes CRX function and induces cone-rod dystrophy in a mouse model of SCA7. Neuron 2001 ; 31 : 913-27.
- 9. Yoo SY, Pennesi ME, Weeber EJ, et al. SCA7 knockin mice model human SCA7 and reveal gradual accumulation of mutant ataxin-7 in neurons and abnormalities in short-term plasticity. Neuron 2003 ; 37 : 383-401.
- 10. Sugars KL, Rubinsztein DC. Transcriptional abnormalities in Huntington disease. Trends Genet 2003 ; 19 : 233-8.
- 11. Helmlinger D, Hardy S, Abou-Sleymane G, et al. Glutamine-expanded ataxin-7 alters TFTC/STAGA recruitment and chromatin structure leading to photoreceptor dysfunction. PLoS Biol 2006 ; 4 : e67.
- 12. Jenuwein T, Allis CD. Translating the histone code. Science 2001 ; 293 : 1074-80.
List of figures
Figure 1
Réorganisation de la chromatine et perte de l’expression des gènes spécifiques des photorécepteurs dans la rétine des souris SCA7.
A. Images de microscopie électronique des noyaux des photorécepteurs de type bâtonnets dans les souris témoins (R7N) et SCA7 (R7E). Les bâtonnets normaux sont caractérisés par une large région d’hétérochromatine centrale et un liseré d’euchromatine périphérique. L’ATXN7 mutée provoque une augmentation très importante du volume nucléaire et une décondensation majeure de l’hétérochromatine centrale. B. Modèle des effets d’une expansion polyglutamine dans ATXN7 (bas). Dans les bâtonnets normaux, les gènes spécifiques des photorécepteurs, tel que le gène codant pour la rhodopsine, sont localisés dans la région périphérique d’hétérochromatine où une forte concentration d’activateurs (A) et de facteurs généraux de la transcription (GTF) permet d’atteindre de forts niveaux d’expression. Dans les souris SCA7, une augmentation du recrutement de TFTC/STAGA sur le promoteur provoque une hyperacétylation des histones. La décondensation de la chromatine entraîne une dilution des facteurs de transcription (A et GTF) expliquant la diminution de l’expression des gènes fortement exprimés dans les bâtonnets (adapté de [11]).