Résumés
Résumé
Cette synthèse traite des procédés d’oxydation avancée (POA) pour le traitement des eaux et des effluents industriels. Ces procédés mettent pour la plupart en combinaison deux ou trois réactifs (oxydants) afin de produire des radicaux hydroxyles. Les radicaux libres sont des espèces hautement actives capables de réagir rapidement et de manière non sélective sur la plupart des composés organiques, réputés difficilement oxydables par voie biologique ou par des traitements chimiques conventionnels. Les POA peuvent être subdivisés en quatre groupes : les procédés d’oxydation chimique en phase homogène (H2O2/Fe2+ et H2O2/O3), les procédés photocatalytiques en phase homogène et/ou hétérogène (H2O2/UV, O3/UV et Fe2+/H2O2/UV; TiO2/UV), les procédés d’oxydation sonochimique et les procédés d’oxydation électrochimique. Le couplage H2O2/Fe2+ représente le système d’oxydation avancée le plus connu et le moins complexe, lequel est souvent employé dans le traitement des effluents industriels. Cependant, dans le domaine de la potabilisation des eaux, le système le plus utilisé et le plus éprouvé est le couplage H2O2/O3 couramment employé pour l’élimination des composés phytosanitaires (pesticides). Les procédés d’oxydation électrochimiques, photocatalytiques et sonochimiques sont des technologies qui nécessitent en général moins de réactif et sont faciles d’automatisation par comparaison aux autres POA. Ces procédés sont présentement en pleine expansion dans le domaine des technologies environnementales, ceci afin d’améliorer les systèmes existants de traitement des eaux usées municipales et industrielles, ou à remplacer les technologies conventionnelles peu efficaces pour l’enlèvement de contaminants organiques réfractaires, inorganiques et microbiens. De nombreuses études réalisées à l’échelle laboratoire ont clairement prouvé l’efficacité des POA pour le traitement de divers effluents. Cependant, le développement de ces procédés dans les filières de traitement des eaux reste encore limité en raison des coûts d’investissement et des coûts opératoires associés. Des solutions et stratégies sont proposées dans ce document, telles que le développement de procédés hybrides et leur couplage avec des traitements biologiques conventionnels, et ce, afin de pallier certaines contraintes spécifiques des POA et faciliter ainsi leur insertion dans les filières de traitement des eaux et des effluents industriels. Ce document a pour objectif de faire une synthèse des différents POA, d’en expliquer leur principe de fonctionnement, de déterminer les différents paramètres les gouvernant, ainsi que leurs applications dans le traitement des eaux et des effluents.
Mots clés:
- Procédé d’oxydation avancée,
- radicaux hydroxyles,
- composé organique réfractaire,
- eau potable,
- effluent industriel
Abstract
This review deals with advanced oxidation processes (AOP) for water and wastewater treatment. Most AOPs combine two or three chemical oxidants in order to produce hydroxyl radicals. These free radicals are species capable of oxidizing numerous complex organic, non-chemically oxidizable or difficulty oxidizable compounds. They efficiently react with carbon-carbon double bonds and attack the aromatic nucleus, which are prevalent features of refractory organic compounds. The AOPs can be divided into four groups: homogenous chemical oxidation processes (H2O2/Fe2+ and H2O2/O3), homogenous/heterogeneous photocatalytic processes (H2O2/UV, O3/UV and Fe2+/H2O2/UV; TiO2/UV), sonification oxidation processes (ultrasound oxidation) and electrochemical oxidation processes. The H2O2/Fe2+ system represents the most common and simplest AOP, which is often employed for the treatment of industrial effluents. However for drinking water treatment, the H2O2/O3 system is commonly used for pesticide removal. Electrochemical, photo-catalytic and sonification oxidation processes require fewer chemicals and are more easily automated than other AOPs. These technologies are effective in improving the treatment of industrial wastes, wastewater and drinking water, for example after their integration into a treatment plant or after their replacement of conventional processes that are found to less effectively eliminate specific organic and inorganic pollutants. The goal of this paper is to review published literature on the use of AOPs for water and wastewater treatment and the removal of refractory pollutants. Specifically, the objectives are: (i) to understand the theory and mechanisms of pollutant removal in AOPs, (ii) to provide a database for AOP applications, and (iii) to suggest new research directions for the development of AOPs.
Key words:
- Advanced oxidation process,
- hydroxyl radical,
- refractory organic compounds,
- drinking water,
- industrial wastewater
Corps de l’article
1. Introduction
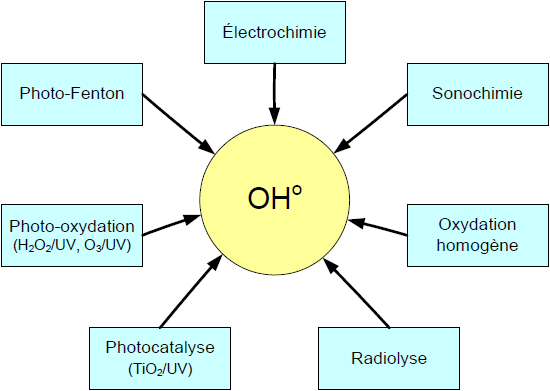
L’industrialisation intensive qui a vu le jour au cours du dernier siècle a causé l’apparition dans l’environnement de polluants émergents réfractaires. Ces substances dites biorécalcitrantes peuvent entraîner une contamination chez les êtres vivants (USEPA, 1997; YONAR et al., 2005). De plus, certains de ces composés sont reconnus pour causer des effets cancérigènes et mutagènes, ou peuvent interférer avec le système hormonal des êtres vivants (perturbateurs endocriniens). Au nombre de ces polluants couramment décelés dans les rejets industriels et urbains, se trouvent des hormones et dérivés hormonaux, des composés phénoliques, des antibiotiques, des organochlorés, des produits cosmétiques etc., lesquels composés sont à l’origine de nombreuses perturbations de la faune aquatique et constituent un risque pour la santé humaine (AURIOL et al., 2007; JÜRGENS et al., 2002; USEPA, 1997). Ces contaminants échappent pour la plupart au traitement classique des eaux usées. La présence de ces polluants émergents dans l’environnement constitue un sujet de préoccupation pour la plupart des agences environnementales des pays industrialisés (Canada, États-Unis et Communauté européenne) (SERVOS et al., 2005; SNYDER et al., 2003a,b; VERSTRAETEN et al., 2003). Dans l’optique de limiter l’arrivée de ces divers types de contaminants réfractaires dans l’environnement, des stratégies de traitement efficaces et écologiques ont été développées. Parmi ces stratégies se trouve l’application des procédés d’oxydation avancée POA. Il s’agit de procédés de traitement oxydatif qui peuvent être regroupés en quatre catégories : i) Procédés d’oxydation chimique en phase homogène (H2O2/Fe2+ et H2O2/O3); ii) Procédés photocatalytiques en phase homogène et/ou hétérogène (H2O2/UV, O3/UV, Fe2+/H2O2/UV et TiO2/UV); iii) Procédés d’oxydation sonochimique, et iv) Procédés d’oxydation électrochimique. Ces technologies sont toutes basées sur la production d’entités radicalaires, notamment, les radicaux hydroxyles (OHo) qui sont des espèces oxydantes les plus puissantes que l’on puisse utiliser dans le domaine du traitement des eaux et des effluents industriels (Figure 1) (SUTY et al., 2003). Par exemple, les radicaux hydroxyles peuvent oxyder un grand nombre de composés organiques avec une vitesse d’oxydation largement supérieure à celle de l’ozone (109 fois plus élevée) (PARSONS, 2004). Les radicaux libres sont des espèces hautement actives susceptibles de réagir avec la quasi-totalité des molécules organiques. Ces composés réagissent avec les doubles liaisons -C–C- et attaquent les noyaux aromatiques, composants majeurs des composés réfractaires (GOGATE et PANDIT, 2004). Ces POA ont été appliqués dans plusieurs secteurs, pour le traitement des eaux de surface et souterraines (PARSONS, 2004), pour l’élimination des odeurs et des composés organiques volatiles (BHOWMICK et SEMMENS, 1994), la décoloration des eaux (HSING et al., 2007; WU et CHANG, 2006), la dégradation de produits phytosanitaires et pharmaceutiques (IKEHATA et EL-DIN, 2006), le traitement des eaux de piscine (PARSONS, 2004), la désinfection des eaux (GONCHARUK et al., 2003), la production d’eau ultra pure (PARSONS, 2004), le traitement des eaux usées industrielles (MARTINEZ-HUITLE et FERRO, 2006), le traitement de lixiviat d’enfouissement et pour le traitement de boues municipales (FLOTRON et al., 2003), etc. Ces techniques peuvent être employées soit comme technique de prétraitement oxydatif conduisant à des composés facilement biodégradables, soit comme méthode de traitement tertiaire pour l’élimination ou la minéralisation complète des polluants résiduels (DROGUI et al., 2007; PANNIZA et al., 2006; SZPYRKOWICZ et al., 2005). La plupart de ces technologies n’ont été développées et appliquées qu’à l’échelle laboratoire, d’autres, par contre, comme le couplage O3/H2O2 ont fait leur preuve à l’échelle industrielle (GALEY et PALAWSKI, 1993).
Figure 1
Différents procédés d’oxydation avancée.
Different advanced oxidation processes..
L’objectif de ce travail est de faire une synthèse sur l’application des procédés d’oxydation avancée (POA) dans le traitement des eaux et des effluents pour l’élimination des polluants réfractaires dans un contexte où les normes environnementales deviennent de plus en plus sévères. De manière plus spécifique, les objectifs sont : i) Comprendre les mécanismes réactionnels des POA, ii) fournir une base de données sur le potentiel d’oxydation de ces technologies tout en indiquant les avantages, les inconvénients et leurs limites dans le traitement des eaux et iii) suggérer de nouvelles directions pour la recherche afin de développer davantage les POA à l’échelle industrielle et commerciale.
2. Radicaux hydroxyles
2.1 Description et caractéristiques de OHo
Le radical hydroxyle (OHo) est une molécule composée d’un atome d’oxygène et d’hydrogène possédant un électron non apparié (électron célibataire) sur son orbital externe. Contrairement aux ions, les radicaux hydroxyles sont produits à partir d’une rupture homolytique d’une liaison covalente, c’est-à-dire que les deux électrons mis en jeu lors de cette liaison sont également partagés, un électron pour chaque atome (MILLET, 1992a,b). Cette caractéristique lui confère un caractère fortement polaire et, par voie de conséquence, il est très réactif vis-à-vis de nombreux composés organiques (aromatiques et aliphatiques), inorganiques et bactériens. Il s’agit d’espèces hautement réactives caractérisées par une demi-vie de l’ordre de 10-9 sec (MAESTRE, 1991; PULGARIN et al., 1994). Son potentiel normal d’oxydoréduction est de 2,81 V par rapport à l’électrode normale à hydrogène. Il est de loin l’un des oxydants les plus puissants qui puissent être utilisés en traitement des eaux (Tableau 1).
Tableau 1
Comparaison des potentiels normaux d’oxydoréduction des principaux oxydants utilisés dans le domaine du traitement des eauxé
Comparison of the oxido-reduction potential of oxidizing agents used in water treatment..
Couple redox |
Réactions |
Potentiel, (V/ENH), 25oC |
Références |
|---|---|---|---|
Cl2/Cl- |
Cl2(g) + 2e → 2Cl− |
1,36 |
DORÉ (1989) |
Br2/Br- |
Br2(l) + 2e → 2Br− |
1,06 |
DORÉ (1989) |
I2/I- |
I2 + 2e → 2I− |
0,53 |
DORÉ (1989) |
OH°/H2O |
OH° + H+ + e → H2O |
2,81 |
Schaller (1996) |
O3/O2 |
O3 + 2H+ + 2e → O2 + H2O |
2,07 |
Guivarch (2004) |
H2O2/H2O |
H2O2 + 2H+ + 2e → 2H2O |
1,77 |
DORÉ (1989) |
MnO4-/Mn2+ |
MnO−4 + 8H+ + 5e → Mn2+ + 4H2O |
1,51 |
DORÉ (1989) |
HClO/Cl- |
HClO + H+ + 2e → Cl− + H2O |
1,49 |
DORÉ (1989) |
ClO2/ClO2- |
ClO2 + e → ClO−2 |
0,95 |
DORÉ (1989) |
S2O82-/SO42- |
S2O2−8 + 2e → 2SO2−4 |
2,05 |
DORÉ (1989) |
2.2 Réactivité des radicaux hydroxyles
Les réactions d’oxydation impliquant les radicaux hydroxyles en présence de substrats organiques (en milieu aqueux) sont principalement des réactions d’addition électrophile et des réactions d’abstraction d’hydrogène (DORÉ, 1989). Les équations (1) et (2) ont été proposées par STRIOLO (1992) pour décrire ces réactions d’addition et d’abstraction :
1er cas : addition du radical OHo sur le composé organique R
2e cas : élimination d’un atome d’hydrogène
Dans les deux cas, des radicaux organiques se forment, lesquels peuvent par la suite réagir avec d’autres radicaux (réaction de terminaison) ou encore réagir avec un autre oxydant moléculaire en solution (réaction de propagation). L’activation initiale de l’oxydant moléculaire conduit à la génération de radicaux hydroxyles (réaction d’initiation). En fait, la génération de ces espèces radicalaires est souvent accompagnée par des réactions en chaîne incluant les étapes d’initiation, de propagation et de terminaison. La réaction de propagation permet de générer de nouvelles espèces radicalaires (ex. HO2o, O22-, Oo, etc.) participant également à l’oxydation des polluants. Cependant, ces espèces intermédiaires sont moins réactives que les radicaux hydroxyles (BIELSKI et al., 1985). Ces réactions en chaîne sont clairement présentées dans les sections 3, 4, 5 et 6 où nous décrivons quelques mécanismes réactionnels des POA. Il faut cependant noter que certains composés organiques possédant des groupements attracteurs d’électrons susceptibles d’appauvrir la densité électronique au point de valence libre, inhibent la propagation de la chaîne d’oxydation induite par les radicaux hydroxyles. Les composés qui satisfont à ce type d’inhibition sont principalement les alcools tertiaires et les esters. MERTZ et WATERS (1949) ont établi une liste des principaux composés aliphatiques, capables ou non d’initier des réactions en chaîne, et définissent un autre groupe de composés réfractaires à l’oxydation par les radicaux OHo à température ambiante (Tableau 2). En effet, le radical OHo ayant un caractère électrophile marqué, les composés substitués par des groupements donneurs d’électrons réagissent plus rapidement et conduisent principalement à la formation de composés ortho ou para hydroxylés. DORÉ (1989) a montré clairement cette différence de réactivité des noyaux aromatiques porteurs d’un groupement donneurs d’électrons (ex. OH, NH2, etc.) comme le phénol et l’aniline par rapport aux cycles porteurs d’un groupement attracteurs d’électrons (–NO2, -COOH, etc.) comme le nitrobenzène ou l’acide benzoïque.
Tableau 2
Oxydabilité des composés organiques par les radicaux hydroxyles (MERTZ et WATERS, 1949).
Organic compound oxidation by hydroxyl radicals (MERTZ et WATERS, 1949)..
Composés oxydables par un mécanisme en chaîne |
|---|
Alcools primaires et secondaires : dérivés méthyl, éthyl, n-propyl, isopropyl |
Hydroxy-acides : acides glycollique, lactique, hydroxybutyrique, thioglycolique |
Éthers : diéthyl éther, dioxane, tétrahydrofuranne, tétrahydrofuranne, tétra hydropyranne |
Aldéhydes : formol, acétaldéhydes |
Acides aminés : glycine, alanine |
Composés oxydables par un mécanisme sans chaîne |
Alcools tertiaires : butanol, alcoolamylique, pinacol, phényldiméthylcarbinol |
Esters : acétate de méthyl, acétate d’éthyle, acétate d’isopropyle |
Acides carboxyliques : acide propionique, butyrique, valérique, succinique, adipique |
Amines : diéthylamines, triéthylamines, pyridine |
Glycols : éthylènes, butylène |
Composés non oxydables par les radicaux OH0 |
Acides carboxyliques : acétique, malonique, maléique, fumarique |
Cétones : acétone, méthyl-éthylène-cétone |
Amides : urée, acétamides |
En particulier, avec le phénol, qui a été le plus étudié, on observe la formation de pyrocathécol ou hydroquinone. Ces produits di-hydroxylés sont à leur tour attaqués par les radicaux libres pour former des composés plus hydroxylés. La réaction subséquente conduit à l’ouverture du cycle aromatique (Figure 2). L’ouverture du cycle aromatique conduit à la formation des principaux produits tels que : l’aldéhyde formique, l’acide maléique, l’acide cétomalonique, l’acide oxalique et l’acide formique (DORÉ, 1989; KARPEL VEL LEITNER et DORÉ, 1997).
Figure 2
Oxydation du phénol par les radicaux hydroxyles (DORÉ, 1989).
Phenol oxidation by hydroxyl radicals (DORÉ, 1989).

2.3 Cinétiques de réaction avec les composés organiques
Ces espèces radicalaires sont peu sélectives vis-à-vis de l’oxydation des polluants, comparativement à la réaction d’oxydation de certains oxydants (ex. O3 et H2O2). Ils réagissent sur les composés organiques avec des constantes cinétiques de réaction pouvant atteindre 109 M‑1 sec‑1 (KARAM et al., 1991). Le tableau 3 compare les constantes cinétiques de l’ozone et des radicaux hydroxyles lors de l’oxydation de quelques molécules organiques. Les radicaux hydroxyles oxydent les molécules comme le benzène, le toluène, le chlorobenzène, le trichloréthylène et le butanol avec une vitesse de dégradation largement supérieure à celle de l’ozone (109 fois plus élevée par rapport à l’ozone). BUXTON et al. (1988) ont largement étudié la cinétique de réaction de OHo sur les composés organiques aliphatiques et aromatiques (Tableaux 4 et 5). D’après les valeurs répertoriées, on peut en déduire que les radicaux hydroxyles réagissent plus vite avec les composés monosubstitués qu’avec les composés polysubstitués. En outre, les radicaux hydroxyles réagissent plus rapidement sur les aromatiques porteurs de groupements activants que ceux présentant des groupements désactivants.
Tableau 3
Comparaison des constantes de vitesse pour l’ozone et les radicaux hydroxyles lors de l’oxydation de quelques molécules organiques (PARSONS, 2004).
Comparison of kinetic rate constants for the production Comparison of kinetic rate constants for the production of ozone and hydroxyl radicals recorded while oxidizing some molecular organic compounds (PARSONS, 2004).
Composés organiques |
Constantes cinétiques (M‑1 sec‑1) |
|
|---|---|---|
O3 |
OHo |
|
Benzène |
2,0 |
7,8 x 109 |
Toluène |
14 |
7,8 x 109 |
Chlorobenzène |
0,75 |
4,0 x 109 |
Trichloroethylène |
17 |
4,0 x 109 |
Tétrachloroéthylène |
<0,10 |
1,7 x 109 |
n-Butanol |
0,60 |
4,6 x 109 |
t-Butanol |
0,03 |
0,4 x 109 |
Tableau 4
Constantes cinétiques de réaction des radicaux hydroxyles en milieu aqueux sur les composés aliphatiques.
Kinetic rate constants of hydroxyl radical production in aqueous media while oxidizing aliphatic compounds.
Composés aliphatiques |
Substituants |
KHO (x 10‑7 M‑1 sec‑1) |
Références |
|---|---|---|---|
Méthane |
‑H |
11 |
Buxton et al. (1988) |
Méthanol |
‑OH |
97 |
Buxton et al. (1988) |
Méthylamine |
‑NH2 |
420 |
Buxton et al. (1988) |
Ion méthyl ammonium |
‑NH3+ |
3,5 |
Buxton et al. (1988) |
Diméthyl éther |
‑OCH3 |
100 |
Buxton et al. (1988) |
Acétone |
‑COCH3 |
11 |
Buxton et al. (1988) |
Acétonitrile |
‑CN |
2,2 |
Buxton et al. (1988) |
Acétamide |
‑CONH2 |
19 |
Buxton et al. (1988) |
Acide acétique |
‑COOH |
1,6 |
Buxton et al. (1988) |
Acétaldéhyde |
‑COH |
73 |
Buxton et al. (1988) |
Dibromométhane |
‑Br2 |
9,0 |
Haag et Yao (1992) |
Trichlorométhane |
‑Cl3 |
5,4 |
Haag et Yao (1992) |
Tableau 5
Composés aromatiques monosubstitués (BUXTON et al., 1988) et polysubstitués (HAAG et YAO, 1992).
Monosubstituted (BUXTON et al., 1988) and polysubstituted (HAAG and YAO, 1992) aromatic compounds.
Composés aromatiques monosubstitués |
Substituants |
KHO (x 10‑9 M‑1 sec‑1) |
Composés aromatiques polysubstitués |
KHO (x 10‑9 M‑1 sec1) |
|---|---|---|---|---|
Benzène |
‑H |
7,8 |
Phtalates |
4,0 |
Phénol |
‑OH |
14 |
Trichlorobenzène |
4,0 |
Aniline |
‑NH2 |
15 |
Dichlorobenzène |
5,0 |
Anisole |
‑OCH3 |
5,4 |
BPCs |
6,0 |
Toluène |
‑CH3 |
3,0 |
HAPs |
1,0 |
Éthylbenzène |
‑CH2CH3 |
7,5 |
Lindane |
5,2 |
Acide benzoïque |
‑COOH |
4,3 |
Atrazine |
2,6 |
Ion benzoate |
‑COO- |
5,9 |
Simazine |
2,8 |
Benzaldéhyde |
‑CHO |
4,4 |
Alschlor |
4,0 |
Acétophénone |
‑C(O)CH3 |
5,9 |
Carbofurane |
7,0 |
Nitrobenzène |
‑NO2 |
3,9 |
Pentachlorophénol |
4,0 |
Benzamide |
‑CONH2 |
3,4 |
Dinoseb |
4,0 |
Chlorobenzène |
‑Cl |
5,5 |
- |
- |
Fluorobenzène |
‑F |
10 |
- |
- |
2.4 Inhibiteurs et promoteurs de radicaux libres
En revanche certains éléments présents dans une eau à traiter pourraient limiter leur production ou leur action : ce sont les inhibiteurs de radicaux (AGUIAR, 1992; AGUIAR et al., 1993; STAEHELIN et HOIGNE, 1985) qui réagissent avec les radicaux hydroxyles OH° sans toutefois générer le radical superoxyde. Dans les eaux naturelles, les principaux inhibiteurs rencontrés sont les carbonates, les bicarbonates et les hydrogénophosphates :
Il est également possible de rencontrer dans les eaux naturelles des espèces appelées promoteurs de radicaux libres (ions formiates, alcools et acides humiques) (XIONG, 1990), qui génèrent un nouveau radical actif (ion radical superoxyde) en consommant un radical hydroxyle :
2.5 Méthodes de détection des radicaux libres
Si le dosage des oxydants moléculaires ne pose pas en général de problème, il n’en va de même pour les oxydants radicalaires (ex. OHo) qui sont très réactifs et dont la durée de vie est extrêmement courte (n’excédant pas le dixième de microseconde). La méthode très répandue de nos jours, mais également très coûteuse, permettant leur analyse directe est la Résonnance de Spin Électronique (RSE). Cette méthode est applicable lorsque les radicaux libres sont produits en quantités relativement élevées. En outre, elle présente des inconvénients comme, par exemple, le fait de devoir effectuer les analyses in situ. Cependant, une autre méthode indirecte de détection des radicaux peut être utilisée. Le principe consiste à piéger le radical étudié par une substance susceptible (ex : p-nitrosodiméthylaniline) de former un adduit stable, lequel sera par la suite isolé et ensuite analysé par une technique de RSE (Résonance de Spin Électronique) (FENG et al., 2003; SCHALLER, 1996).
3. Procédés d’oxydation chimique en phase homogène
3.1 Réactif de Fenton : couplage H2O2/Fe2+
3.1.1 Mécanisme réactionnel
Le système d’oxydation radicalaire le plus connu est le réactif de Fenton. Le traitement chimique par réactif de Fenton est une technique qui consiste à initier des réactions de décomposition du peroxyde d’hydrogène (H2O2) par des sels métalliques (en particulier des métaux de transition) en vue de générer des espèces radicalaires (OHo, HO2o, etc.) très réactives vis-à-vis des polluants organiques (LIN et LO, 1997). Ces ions métalliques peuvent être présents dans l’effluent à traiter, sous forme libre ou le plus souvent complexée. La décomposition de H2O2 par les ions ferreux a été initialement proposée par HABER et WEISS (1934). La réaction est du type radicalaire :
Pour de faibles rapports H2O2/Fe2+ et en milieu suffisamment acide (0 < pH < 3), les réactions d’initiation (7) et de terminaison (9) sont prédominantes. L’augmentation du rapport H2O2/Fe2+ favorise, par contre, la réaction de propagation (8) et la formation de radical hydroperoxyle HO2° capable de réduire ensuite le Fe3+ en Fe2+ et de propager le cycle de décomposition (DENG et ENGLEHARDT, 2006). La décomposition du peroxyde d’hydrogène peut être également réalisée par les ions ferriques suivant la réaction :
Plusieurs métaux sont susceptibles d’initier des réactions de décomposition du peroxyde d’hydrogène (COLOBERT et al., 1962; MAESTRE, 1991).
Dans cette équation, (n) représente le degré d’oxydation du métal (M). Les métaux susceptibles de favoriser la décomposition radicalaire du peroxyde d’hydrogène sont Fe, Cr (VI), Ti, Cu, Mn, Co(I), Al, etc. Les catalyseurs les plus utilisés restent le cuivre et l’aluminium (BARB et al., 1951).
3.1.2 Application du réactif de Fenton dans le traitement des eaux et des effluents
Généralement, le procédé Fenton est composé de quatre étapes principales : ajustement du pH, oxydation, neutralisation et coagulation/précipitation (Figure 3). Le défaut majeur du procédé Fenton conventionnel est qu’il nécessite, pour être efficace, une zone de pH allant de 2,0 à 4,0, avec une valeur optimale à pH 2,8 déterminée par PIGNATELLO (1992). Au‑delà des valeurs de pH comprises entre 3,0 et 4,0, le fer (Fe2+ et Fe3+) est susceptible de précipiter et former des hydroxydes de fer, induisant ainsi une faible activité catalytique. Contrairement aux autres POA, le procédé Fenton génère dans certains cas des résidus métalliques (hydroxyde de fer) qu’il faut éliminer, ce qui représente un coût supplémentaire. Dans l’optique d’y remédier, il est possible d’utiliser des ligants organiques permettant, par complexation, de limiter la perte de fer sous forme d’hydroxyde et de travailler à pH plus élevé tout en conservant l’activité catalytique du fer. Cependant, l’utilisation de ligants organiques implique d’autres inconvénients, car ceux-ci peuvent réagir avec les radicaux hydroxyles formés. De plus, le fer étant sous forme de complexe, il sera plus difficile de l’éliminer par précipitation. L’ajout de complexant génèrera évidemment un coût supplémentaire. L’activation du peroxyde d’hydrogène par les ions ferreux est souvent utilisée pour la dégradation de certains composés organiques réfractaires. Par exemple, GOEL et al. (2003) ont étudié l’efficacité du réactif de Fenton (H2O2/Fe2+) pour la dégradation du naphtalène présent dans les eaux brutes destinées à la consommation humaine. Les essais ont été effectués à différents pH (pH 4,0 et pH 7,0) en utilisant l’action combinée peroxyde/fer ferreux en imposant différentes concentrations de réactifs, ou encore, en utilisant l’action seule du peroxyde. Les meilleurs résultats ont été enregistrés grâce à l’action combinée peroxyde/fer ferreux. Pour des concentrations relativement élevées de H2O2 (10 à 12 mg H2O2•L‑1), le peroxyde d’hydrogène en présence de fer ferreux (30 mg Fe2+•L‑1) permettait d’obtenir des taux d’élimination de naphtalène de 99 % et 96 % à des pH respectifs de 4,0 et 7,0. En comparaison, pour des concentrations relativement faibles de H2O2 (2 à 3 mg H2O2•L‑1), des rendements de dégradation de 84 et 68 % étaient respectivement enregistrés à pH 4,0 et 7,0. L’action seule du peroxyde d’hydrogène était quasiment inefficace pour l’oxydation du naphtalène.
Figure 3
Traitement d’effluent par le procédé Fenton (GOGATE et PANDIT, 2004)
Effluent treatment using the Fenton process (GOGATE and PANDIT, 2004)

D’autres études ont également montré l’efficacité du procédé Fenton dans de multiples applications. Par exemple, le réactif de Fenton a été utilisé pour la décoloration d’effluents provenant d’industries de teinture (KUO, 1992), pour la destruction de composés organiques toxiques tels que le 2,4,6-trinitrotoluène (LI et al., 1997a, b), le 2,4-dinitrophénol (KANG et al., 1999; WANG et al., 1999), le chlorobenzène (WATTS et al., 1997), le tétrachloroéthylène (YOSHIDA et al., 2000), les chlorophénols (BARBENI et al., 1987; POTTER et ROTH, 1993; WATTS et al., 1990), les haloalcanes (TANG et TASSOS, 1997).
Une autre approche de l’application du réactif de Fenton, appelée procédé électro-Fenton, a été développée (BRILLAS et CASADO, 2002; BRILLAS et al., 1999; DURAN MORENO et al., 2004; OTURAN et al., 1999). Le procédé consiste à produire in situ et par voie électrolytique les réactifs (Fe2+ et H2O2) impliqués dans le réactif de Fenton, de sorte que le procédé ne requiert aucun ajout de produits supplémentaires (DURAN MORENO et al., 2004). Le peroxyde d’hydrogène est produit par réduction cathodique de l’oxygène, alors que les ions ferreux sont produits en solution par dissolution anodique d’une électrode de fer. Par ailleurs, une autre application du réactif de Fenton consiste à générer in situ le H2O2 par voie électrochimique. Le fer ferreux est alors initialement injecté dans le système avec la possibilité de le régénérer électrochimiquement par réduction du fer ferrique à la cathode (MATSUE et al., 1981; OTURAN et PINSON, 1992). Dans ce procédé, les quantités de sels de fer sont considérablement réduites puisque le catalyseur de fer ferreux est continuellement régénéré à la cathode. Les polluants organiques peuvent alors être détruits par les radicaux hydroxyles produits par la réaction de Fenton, mais également par les radicaux HO2o formés par oxydation de H2O2 à l’anode (BRILLAS et al., 1995). La réaction de Fenton électrochimiquement assistée a déjà été appliquée avec succès pour la dégradation et la minéralisation de nombreux composés organiques variés tels que les chlorophénols, les produits phytosanitaires (insecticides, herbicides, fongicides), les polluants industriels (nitrophénols), benzène (BRILLAS et al., 1995; GÖZMEN et al., 2003; OTURAN et al., 2001).
3.2 Procédé de peroxonation H2O2/O3
Le principe du procédé de peroxonation repose sur le couplage entre l’ozone et le peroxyde d’hydrogène afin de produire des radicaux libres. Ce procédé est plus efficace que l’ozonation seule, car le peroxyde d’hydrogène a pour but d’accélérer la décomposition de l’ozone dans l’eau et ainsi produire beaucoup plus de radicaux hydroxyles.
3.2.1 Mécanisme réactionnel du système de peroxonation
Le peroxyde d’hydrogène réagit très rapidement sous sa forme ionisée (HO2-; pKa = 11,6) sur l’ozone pour former des radicaux libres (OHo, HO2o) selon la réaction suivante (PAILLARD et al., 1988) :
Une fois libérés lors de la réaction de HO2- avec l’ozone, ces radicaux initient ensuite d’autres mécanismes radicalaires de décomposition du peroxyde d’hydrogène (Figure 4). Ces réactions ont été mises en oeuvre de manière optimale par PAILLARD et al. (1988). Le pH est alors de 7,7 et le rapport H2O2/O3 est égal à 0,5.
Figure 4
Mécanisme d’activation du peroxyde d’hydrogène par l’ozone (DORÉ, 1989).
Activation mechanism of H2O2 by ozone, (DORÉ, 1989).

3.2.2 Application du procédé de peroxonation
Le peroxyde d’hydrogène couplé à l’ozone est performant pour éliminer les micropolluants ou composés toxiques (pesticides, hydrocarbures, etc.) présents dans les eaux potables, les eaux résiduaires industrielles ou les nappes phréatiques (CHROMOSTAT et al., 1993; PAILLARD, 1994). Ce couplage peut éventuellement être utilisé pour assurer un effet rémanent surtout en traitement de potabilisation à condition d’utiliser un excès de peroxyde. L’oxydation par O3/H2O2 est insérée entre la filtration sur sable et la filtration sur charbon actif en grain (CAG). Le traitement a pour objectifs principaux d’abaisser la teneur en micropolluants (les pesticides en particulier) avant la filtration sur CAG, pour augmenter la durée de vie du CAG. L’oxydation est généralement effectuée avec des temps de séjour de l’eau dans les contacteurs d’ozone, de l’ordre de 10 min, avec une injection fractionnée à divers niveaux du contacteur d’ozonation afin de maintenir le rapport O3/H2O2 optimal en tout point du réacteur. Il est à noter que la concentration résiduelle de H2O2 ne peut pas dépasser la valeur maximale de 0,5 mg•L‑1 autorisée dans l’eau traitée. Ce système est affecté par les mêmes contraintes que l’ozonation seule, à savoir la faible solubilité de O3, la forte consommation énergétique et la sensibilité à de nombreux paramètres tels que le pH, la température, le type de polluant et les réactions parasites consommant les OH° (défaut commun à tous les POA) (BUXTON et al., 1988; HERNANDEZ et al., 2002). L’avantage du système réside en sa simplicité d’utilisation et son efficacité bactéricide, ce qui explique son développement à grande échelle aux États-Unis et en Europe comme étape de désinfection dans le traitement des eaux potables.
L’ozonation couplée au peroxyde d’hydrogène est un procédé qui a fait ses preuves au niveau industriel. GALEY et PASLAWSKI (1993) décrivent les conditions de mise en oeuvre de ce système appliqué dans trois usines des Syndicats des Eaux d’Ile-de-France (Choisy-le-Roi, Neuilly-sur-Marne et Méry-sur-Oise) pour l’élimination des pesticides (atrazine, simazine, et terbutryne), des phényl-urées et des organochlorés (lindane et endosulfan). Les essais ont été effectués dans des usines pilotes fonctionnant avec des cuves d’ozonation constituées de trois à quatre compartiments (deux compartiments de transfert et deux compartiments de contact) avec des débits allant jusqu’à 10 000 m3•j‑1. Des taux d’ozonation variant entre 3,5 et 4,5 g•m‑3 ont été appliqués, alors qu’un ratio de 0,4 de H2O2/O3 (g•g‑1) était imposé au cours du traitement. L’eau était dopée en pesticide à l’entrée du pilote à une concentration de 1,0 µg•L‑1 environ. Dans tous les cas, le respect de la norme européenne (0,1 µg•L‑1 pour chaque substance individualisée) était assuré en sortie de l’usine. Les triazines (atrazine, simazine et terbutryne) étaient éliminées à plus de 80 % par le traitement O3/H2O2, l’atrazine étant la triazine la plus difficile à traiter. De même, les phényl-urées étaient très réactives à l’ozone seule ou couplée à H2O2 (plus de 90 % d’élimination). En revanche, les organochlorés (lindane et endosulfan) étaient mal éliminés en présence de H2O2 (environ 30 % d’élimination).
4. Procédés photocatalytiques en phase homogène
Les technologies photochimiques sont simples d’utilisation, propres et peu coûteuses. De plus, elles ont la particularité de pouvoir à la fois désinfecter et traiter les contaminants. Ainsi, au cours de ces dernières années, elles sont devenues très attractives et sont susceptibles de remplacer des techniques de traitement conventionnelles telles que l’adsorption sur charbon actif et la biodégradation. Le couplage des radiations UV avec des oxydants puissants tels que l’ozone et le peroxyde d’hydrogène permet de dégrader les polluants de trois manières différentes : i) photo-dégradation (photo-dissociation) utilisant les rayons UV pour exciter les molécules polluantes et les dégrader, ii) oxydation par action directe des oxydants O3 et H2O2 et iii) oxydation par photolyse de l’oxydant induisant la formation de radicaux libres hydroxyles.
4.1 Couplage peroxyde/ultraviolet (H2O2/UV)
La photolyse du peroxyde d’hydrogène, à des longueurs d’onde comprises entre 200 et 300 nm, provoque une coupure homolytique de la liaison O — O de la molécule de H2O2 et conduit à la formation de radicaux hydroxyles qui participent également, par des réactions secondaires, à la décomposition du peroxyde d’hydrogène (BUXTON et al., 1988, cité par CRISSOT, 1996) :
La vitesse de production des radicaux libres dépend de plusieurs facteurs (CRISSOT, 1996) dont le pH, les caractéristiques des lampes UV (spectre d’émission, énergie), et les caractéristiques du milieu (facteur de transmission du rayonnement UV, turbidité). La réaction est plus rapide en milieu basique. Pour des pH inférieurs à 10, les vitesses sont faibles. Ce phénomène s’explique par une plus grande absorption de la radiation UV par l’anion hydroperoxyde. Ce procédé est souvent utilisé pour la décontamination des eaux souterraines (ECKENFELDER et al., 1992), pour l’élimination des cyanures présents dans les nappes phréatiques, et pour l’élimination des polluants tels que trichloréthylène (TCE), tétrachloréthylène, benzène etc. (DORÉ, 1989). Cette association permet également d’assurer un effet rémanent dans le cas d’une décontamination bactérienne.
4.2 Couplage ozone/ultraviolet (O3/UV)
L’ozone en solution aqueuse absorbe les radiations UV avec un maximum d’adsorption à 253,7 nm (VAN CRAEYNEST et al., 2004). Cette réaction conduit à la production de radicaux libres qui sont des espèces oxydantes très réactives et efficaces pour éliminer les polluants organiques et bactériens.
Ce type de couplage a pour la plupart été employé pour l’élimination des composés organochlorés volatils (COCV). BHOWMICK et SEMMENS (1994) ont étudié la performance du couplage O3/UV pour l’oxydation de plusieurs COCV (CHCl3, CCl4, TCE, PCE, 1,1,2-TCA etc.). Ils constatèrent que l’ozonation directe contribuait à l’oxydation des CHCl3, alors que la réaction par les radicaux hydroxyles (OHo) contribuait à l’oxydation de CHCl3, 1,1,2‑TCA et, dans une moindre mesure, à l’élimination des TCE. Cependant, ni l’ozone ni les radicaux hydroxyles n’étaient à même de fournir des résultats satisfaisants concernant l’élimination de CCl4. Par ailleurs, STRIEBIG et al. (1996) ont étudié également le couplage UV/O3 à l’aide d’un réacteur pilote hybride comportant un système de contrôle de la pollution de l’air. Ils observèrent une dégradation significative de TCE, alors que très peu ou aucun effet n’avait été observé sur les composés non chlorés organiques volatils. Par ailleurs, ce couplage (O3/UV) a également été étudié pour le traitement d’effluents contenant divers types de polluants organiques tels que pesticides (IKEHATA et al., 2006; LAFI et AL-QODAH, 2006), perturbateurs endocriniens (IRMAK et al., 2005; LAU et al., 2007), composés pharmaceutiques (GEBHARDT et SCHROEDER, 2007), surfactants (AMAT et al., 2007), colorants (HSING et al., 2007; WU et CHANG, 2006; YONAR et al., 2005; SHU et CHANG, 2004), nitrobenzène (TONG et al., 2005), antibiotiques (AKMEHMET et OTKER, 2004).
4.3 Couplage réactif de Fenton/ultraviolet (Fe2+/H2O2/UV)
Le procédé Fenton, décrit précédemment, utilise le fer comme catalyseur pour ainsi produire, par l’intermédiaire du peroxyde d’hydrogène, des radicaux hydroxyles. Le procédé photo-Fenton est un POA hybride qui utilise une source d’irradiation, généralement un rayonnement UV, pour augmenter le taux de radicaux libres en stimulant la réduction du Fe3+ en Fe2+. Lors de ce procédé, l’irradiation possède une double fonctionnalité. Celle-ci permet de décomposer par photolyse le peroxyde d’hydrogène pour former deux radicaux hydroxyles (cf., système H2O2/UV) mais aussi d’augmenter l’activité catalytique du fer.
En fait, la réaction limitant l’action catalytique du fer dans le procédé conventionnel de Fenton (sans irradiation) est la réaction de réduction de Fe3+ (réaction plus lente). Ainsi, d’après la réaction ci-dessus, l’irradiation permet de régénérer plus facilement Fe2+ sans consommer du peroxyde d’hydrogène, tout en formant un radical hydroxyle. Le couplage du procédé Fenton à une source d’irradiation permet d’augmenter considérablement la production de radicaux libres et, ainsi, augmenter le taux de dégradation des contaminants organiques (SUN et PIGNATELLO, 1993).
4.4 Application des procédés photocatalytiques en phase homogène
Le couplage des radiations UV avec le peroxyde d’hydrogène ou l’ozone apporte un bénéfice dans le traitement de contaminants (composés persistants) qui nécessite des conditions d’oxydation élevées. La concentration en peroxyde d’hydrogène doit en outre être optimisée. L’efficacité de ces procédés dépend de la concentration en réactifs (O3 et H2O2) et des longueurs d’onde appliquées (SAULEDA et BRILLAS, 2001). Comme on a pu le voir, l’utilisation de radiations UV permet d’augmenter sensiblement le taux de dégradation des composés organiques. Néanmoins, cela ajoute au procédé des inconvénients liés à l’irradiation. En effet, il faut tenir compte des caractéristiques spectrales et de la puissance des lampes utilisées, de leur encrassement, des types de polluants présents (absorption et rendement quantique), de la turbidité des effluents limitant le parcours des rayonnements ainsi, évidemment, que des coûts d’installation et de fonctionnement supplémentaires.
Les procédés de photo-oxydation ont été fréquemment étudiés pour le traitement d’effluents issus d’industries textiles (KANG et al., 1999, 2000; PRAKASH et al., 2007; XU et al., 2008), de pâte de papier (HELMY et al., 2003; KULKARNI et al., 2006; MOISEEV et al., 2004) et d’extraction d’huile d’olive (AZABOU et al., 2007; MIRANDA et al., 2001; NAJJAR et al., 2007). Dans le cas d’effluents issus des industries textiles, ces études se sont focalisées principalement sur la décoloration des eaux, l’élimination de la toxicité et l’amélioration de la biodégradabilité. Les différents colorants et teintures utilisés dans ce type d’industrie se retrouvent dans les eaux usées et absorbent fortement les radiations UV, ce qui nécessite une augmentation significative de la quantité de réactifs et de l’intensité lumineuse. Le traitement par oxydation photochimique utilisé seul ne parait pas économiquement suffisant pour être étendu à l’échelle industrielle. EL-DEIN (2002) a montré que la combinaison d’un traitement d’oxydation H2O2/UV avec un traitement biologique offrait une alternative plus intéressante et économique. Les procédés photocatalytiques en phases homogènes présentent cependant des avantages à dégrader complètement les composés organiques persistants en dioxyde de carbone, eau et sel inerte. Néanmoins, la faisabilité économique reste un des facteurs qui limite cette technique. Pour cette raison, de nombreuses recherches sont en cours afin de coupler ces techniques d’oxydation photochimique avec d’autres procédés tels que la coagulation chimique, les techniques membranaires, la filtration sur charbon actif et le traitement biologique. Ce type d’approche pourrait résoudre ainsi les limitations liées à ce traitement d’oxydation, par exemple : la forte turbidité, la présence de particules solides, de métaux lourds, la forte DCO et DBO, et contribuerait sans doute à diminuer les coûts de traitement associés.
5. Procédés photocatalytiques en phase hétérogène
5.1 Réactivité des procédés photocatalytiques
La photocatalyse hétérogène est une technologie basée sur l’irradiation d’un catalyseur, en général un semi-conducteur (ex. TiO2), qui peut être photo-excité pour former des sites électro-donneurs ou électro-accepteurs d’électrons et ainsi induire des réactions d’oxydoréduction. En effet, si les photons absorbés possèdent une énergie supérieure au gap d’énergie entre la bande de valence et la bande de conduction, des paires d’électron-trou se créent dans le semi-conducteur (des trous dans la bande BV et des électrons dans la bande BC), ce qui correspond au schéma du principe montré dans la figure 5.
Figure 5
Photo-excitation d’un semi-conducteur
Photoexcitation of a semiconductor.

Ces paires électron-trou peuvent se recombiner pour libérer de la chaleur ou migrer à la surface du semi-conducteur et, ainsi, réagir avec les espèces adsorbées à la surface. Parmi les différents semi-conducteurs photocatalytiques, le dioxyde de titane (TiO2) semble particulièrement bien adapté au traitement des eaux, car il est chimiquement et biologiquement inerte, facile à produire et à utiliser, peu coûteux, et actif d’un point de vue photocatalytique.
De plus, il s’agit d’un des rares semi-conducteurs possédant un gap d’énergie pouvant être compatible à celui fourni par les photons solaires (EgapTiO2 = 3,2 - 3,0 V), bien que la fraction d’énergie solaire absorbée soit de l’ordre de 6,0 % (UV < 380 nm). Suite à l’irradiation du semi-conducteur, les photo-trous formés peuvent oxyder directement le contaminant adsorbé ou alors oxyder les groupes hydroxyles à la surface de TiO2 (Ti4+OH) pour former des radicaux hydroxyles (Ti4+OH+o). La dégradation de polluants contenus dans les eaux usées peut ainsi s’effectuer directement à la surface du semi-conducteur ou indirectement en interagissant avec des radicaux hydroxyles. L’oxydation indirecte par les radicaux libres reste la voie de dégradation prédominante. De plus, si du peroxyde d’hydrogène ou de l’ozone est ajouté dans le réacteur, l’irradiation pourra servir également à la photolyse de ces oxydants et, ainsi, produire plus de radicaux hydroxyles.
L’utilisation du catalyseur lors du processus photocatalytique peut s’effectuer de deux manières différentes, soit sous une forme dispersée, ou soit sous une forme fixe (support catalytique). L’avantage du catalyseur sous forme dispersée réside dans sa simplicité d’utilisation et sa surface spécifique importante. De plus, une aération peut être couplée au processus pour limiter la recombinaison des paires électrons-trous et favoriser la dispersion du catalyseur. Cependant, les performances du réacteur peuvent être affectées à cause d’une faible efficacité d’irradiation liée à l’opacité de la « boue » catalytique, ainsi qu’à l’encrassement de la source lumineuse. Les réacteurs utilisant un catalyseur fixe n’ont pas ce genre de contraintes et n’exigent pas de séparation des fines particules catalytiques en fin du procédé, mais sont soumis à la nécessité d’obtenir une couche catalytique stable et active. La quantité et le type de catalyseur dépendent de la source d’irradiation, du type et de la concentration du polluant, ainsi que du réacteur. Le réacteur doit être conçu de telle sorte que l’irradiation soit uniforme sur la surface entière du catalyseur. De plus, pour la conception de réacteurs à grande échelle, l’élimination de la résistance liée au transfert de masse doit être prise en compte. Le pH optimal du milieu dépend fortement du type de polluant. Ainsi, pour des polluants faiblement acides, le rendement photocatalytique augmente en faisant baisser le pH car cela diminue sa polarité et ainsi facilite son adsorption sur le catalyseur, et inversement pour des polluants faiblement basiques. Il est à noter par ailleurs que lors du processus d’oxydation photochimique, les irradiations émises peuvent directement conduire à une transformation du polluant cible. Par exemple, le polluant organique (R) est d’abord photo-excité (28), lequel pourra par la suite réagir avec l’oxygène dissous dans l’eau (29) et conduire à la formation de sous-produits (30).
5.2 Application au traitement d’effluents
L’intérêt principal de la photocatalyse réside dans son efficacité à éliminer une large gamme de polluants, en particulier ceux difficilement oxydables par les méthodes conventionnelles (Tableau 6). En outre, cette technique permet de transformer certains composés minéraux toxiques en composés moins toxiques. Il est en effet possible d’oxyder les anions, nitrite, sulfite, cyanure et bromate en d’autres composés inoffensifs tels que NO3-, SO42-, CO2 et Br‑, respectivement (MILLS et LE HUNTE, 1997). La photocatalyse permet également, par l’intermédiaire des radicaux hydroxyles, de détruire les polluants biologiques pathogènes tels que les virus, bactéries et moisissures. Dans le cas d’un effluent fortement chargé en matière organique (ex : COD > 800 mg•L‑1), l’absorption trop importante de l’intensité de la radiation rend la technique inadaptée, sauf pour un traitement final.
Tableau 6
Exemples de composés organiques pouvant être photominéralisés par le TiO2 (MILLS et LE HUNTE, 1997).
Examples of organic compounds that may be photomineralized by TiO2 (MILLS and LE HUNTE, 1997).
Classes |
Exemples |
|---|---|
Alcanes |
Méthane, isobutane, heptane, cyclohexane, paraffine |
Haloalcanes |
Mono-, di-, tri-, tétrachlorométhane, tribromoéthane |
Alcools |
Méthanol, iso-propyl alcool, glucose, sucrose |
Acides carboxyliques |
Formique, éthanoïque, diméthyléthanoique, oxalique |
Alcènes |
Propène, cyclohexène |
Haloalcènes |
Perchloroéthène, dichloroéthène, 1,1,2-trichloroéthène |
Aromatiques |
Benzène, naphtalène |
Haloaromatiques |
(Di)-chlorobenzène, bromobenzène |
Nitrohaloaromatiques |
3,4-dichloronitrobenzène |
Phénols |
Phénol, hydroquinone, catéchol, méthylcatéchol, crésol |
Halophénols |
Pentachlorophénol, fluorophénol, difluorophénol |
Acides carboxyliques aromatiques |
Benzoïque, aminobenzoïque, phtalique, salicylique, hydroxybenzoïque, chlorohydroxybenzoïque |
Polymères |
Polyéthylène, PVC |
Surfactants |
SDS, polyéthylène glycol, dodécyl benzène, sulfonate, triméthylphosphate, tétrabutyl ammonium phosphate |
Herbicides |
Méthyl viologen, atrazine, simazine, prometron, propetryne et bentazon |
Pesticides |
DDT, parathion, lindane |
Colorants |
Bleu de méthylène, rhodamine B, orange de méthyle, fluoresceine |
6. Procédés d’oxydation sonochimique
6.1 Mécanisme réactionnel des ultrasons en milieu aqueux
Les ultrasons sont de plus en plus utilisés en milieu industriel pour notamment accélérer ou activer un mécanisme réactionnel. L’intérêt des ultrasons réside dans leur aspect non polluant et leur facilité d’automatisation. En milieu aqueux, les ultrasons peuvent agir suivant deux modes d’action, soit par action chimique (indirecte), ou encore par une action physique (directe). L’action indirecte, souvent obtenue à haute fréquence, conduit à la génération de radicaux libres (HOo, HO2o et Oo) induite par rupture homolytique de la molécule d’eau ou d’oxygène (LORIMER et MASON 1987; RIEZ et al., 1985; TRABELSI et al., 1996). Aussi, à haute fréquence, les ultrasons peuvent provoquer la formation de peroxydes dans l’eau. Ainsi, les ultrasons peuvent s’appliquer à l’éradication des pathogènes et à l’élimination des composés responsables des odeurs malodorantes présents dans les boues d’épuration (NAFFRECHOUX et al., 2003). L’action directe induite par les ultrasons est également intéressante pour le traitement d’effluents industriels. En effet, les ultrasons génèrent des bulles de cavitation, créant ainsi des micro-jets de liquide et des forces de cisaillement très puissants lors de leur implosion, lesquels sont favorables à une meilleure homogénéisation du milieu réactionnel. Aussi, les ultrasons peuvent avoir dans l’eau des effets biocides induits par ces phénomènes de cavitation qui brisent les cellules bactériennes (HUA et HOFFMANN, 1997). Lorsqu’une bulle de cavitation s’effondre (implosion), cela entraîne une élévation locale de température de plusieurs milliers de degrés (2 000 à 5 000 °C) et une pression de plusieurs centaines d’atmosphère (500 atm). Dans ces conditions extrêmes, des radicaux issus de la sonolyse de l’eau sont alors éjectés dans le milieu où ils vont réagir avec les composés en solution. Les bulles de cavitation agissent alors comme des microréacteurs. En effet, lors du phénomène de cavitation, la destruction/oxydation des polluants peut se faire de deux manières différentes, soit par attaque des radicaux libres, ou soit par pyrolyse. Les mécanismes contrôlant la destruction dépendent de la nature du polluant, mais aussi de l’intensité de la cavitation, soumise elle-même aux conditions opératoires du réacteur sonochimique. Le processus de cavitation utilisé en traitement des eaux est réalisé en utilisant des ondes sonores de fréquences variant entre 16 kHz et 3 MHz.
6.2 Application des ultrasons au traitement d’effluents
Le processus de cavitation dépend de plusieurs paramètres tels que la fréquence ultrasonore, l’intensité, la composition de l’effluent et le type de réacteur. Le phénomène de cavitation repose sur deux composantes essentielles, le milieu liquide et la source de vibrations de haute énergie. Le milieu liquide est nécessaire car la sonochimie est dirigée par la cavitation acoustique se produisant uniquement dans un liquide. La fréquence et l’amplitude des ondes ultrasonores générées par l’émetteur (sonde ultrasonore) sont des paramètres déterminants pour l’efficacité de dégradation des polluants. Les valeurs optimales de fréquence et d’amplitude, afin d’avoir la meilleure efficacité de traitement, dépendent des caractéristiques de l’effluent à traiter (type de polluants, concentration, etc.) ainsi que des conditions opératoires (volume du réacteur, temps de traitement, etc.) (GOGATE et PANDIT, 2004). L’objectif, dans le cas du traitement des eaux usées contenant des composés persistants, est de produire un maximum de radicaux libres OHo. Des études (RIEZ et al., 1985) ont montré qu’à fréquence peu élevée (100 à 20 kHz), l’intensité de la cavitation augmente impliquant alors une plus grande formation de radicaux libres et donc une meilleure efficacité de dégradation des composés à l’intérieur de la bulle de cavitation. A contrario, la durée de vie des bulles de cavitation augmente et donc une certaine proportion des radicaux libres formés se recombinent pour former de l’eau et n’ont donc pas le temps d’être efficaces. GOGATE et PANDIT (2001) ont observé une meilleure efficacité des sondes ultrasoniques ayant une grande surface de contact car elles offrent une dissipation énergétique plus uniforme. Les propriétés physicochimiques de l’effluent (pression de vapeur, tension superficielle, viscosité, présence d’impuretés/gaz, etc.) affectent de manière importante les performances du processus sonochimique. En effet, la taille et le nombre des bulles de cavitation produites dépendent des propriétés du liquide. L’idéal est de minimiser la puissance nécessaire au déclenchement du phénomène de cavitation (en modifiant les propriétés physico-chimiques) pour que l’énergie nécessaire à la croissance et à l’effondrement des bulles de cavitation soit maximale, ce qui induirait une augmentation de la pression d’implosion (MASON 1990, GOGATE et PANDIT 2000a). En outre, la dégradation sonochimique des polluants est plus efficace à faible concentration. Dans le cadre de traitement des eaux usées, s’il semble difficile de modifier leurs propriétés physicochimiques, il est cependant envisageable d’effectuer un traitement préalable afin d’en éliminer une partie. De nombreuses études ont montré l’efficacité des procédés d’oxydation sonochimique pour dégrader certains composés organiques présents dans les eaux usées. BERLAN et al. (1994) ont efficacement éliminé le phénol à la fréquence de 541 kHz en formant par oxydation des composés intermédiaires (hydroquinone et catéchol) puis en attaquant ceux-ci jusqu’à minéralisation complète. De même, PETRIER et al. (1999) ont eu la même approche avec le 4-chlorophénol à 500 kHz conduisant à la formation de sous-produits caractéristiques de l’oxydation par les radicaux libre OHo (ex : le 4-chlorocatéchol), suivie d’une dégradation finale (CO, CO2 et HCOOH).
Dans le but de minimiser cette dépense énergétique, les ultrasons sont très souvent utilisés en synergie avec d’autres procédés. Utilisés en combinaison avec des oxydants chimiques (procédés chimiques), ils contribuent efficacement à la désinfection avec des quantités plus faibles de réactifs et des temps plus courts (BERGER et MARR, 1960; SHANER, 1964). Ils permettent de casser certains agrégats et d’éliminer par la suite les microorganismes. Ils permettent de désolidariser les bactéries des solides ou des agglomérats bactériens, lesquels assurent leur protection (DUGUET et CARPENTIER, 1989). Aussi, couplés avec des techniques électrochimiques (procédé sono-électrochimique), ils permettent d’activer la surface des électrodes et d’augmenter considérablement le transfert de matière à l’interface électrode-electrolyte. Aussi, ils permettent d’éliminer les bulles de gaz (notamment O2 et H2) et les impuretés ou débris fixés à la surface des électrodes. Ceci a pour avantage, d’une part, d’éviter l’encombrement des électrodes et, d’autre part, de minimiser la chute ohmique, favorisant ainsi une baisse de la consommation énergétique (TRABELSI et al., 1996).
7. Procédés d’oxydation électrochimique
L’électrochimie est principalement basée sur un transfert d’électrons, ce qui la rend particulièrement intéressante du point de vue environnemental et peut s’appliquer sur un grand nombre de polluants. L’électrochimie a prouvé son efficacité pour le traitement d’effluents toxiques contenant des cyanures ou pour la récupération de métaux lourds par réduction cathodique. En outre, elle peut également traiter des solutions aqueuses chargées en matière organique, soit sous forme d’un prétraitement pour transformer les composés réfractaires en produits biodégradables, ou soit en traitement final pour minéraliser complètement les composés organiques. L’oxydation électrochimique présente l’avantage de pouvoir réagir sur les polluants de deux manières différentes, soit de manière directe ou indirecte.
7.1 Oxydation anodique directe
Les effluents industriels et les eaux usées municipales peuvent être ainsi traités par action directe du courant électrique au cours de l’électrolyse. Il y a effet direct sur une espèce en solution lorsque l’échange d’électrons s’effectue exclusivement sur l’électrode. Des électrodes insolubles dites à forte surtension d’oxygène (Pt, Ti/IrO2; Ti/RuO2; Ti/SnO2, PbO2, Gr, etc.) sont couramment utilisées (GANDINI et al., 1998; RAJESHWAR et IBANEZ, 1997). Employées à l’anode, ces électrodes permettent l’oxydation complète d’un grand nombre de molécules organiques contenues dans les eaux usées. Les sous-produits résultant de l’oxydation anodique sont l’acide carbonique, l’eau et l’ammoniaque et d’autres produits. Dans bien des cas, l’oxydation anodique de polluants organiques ou inorganiques conduit à la formation de composés plus simples, non toxiques ou moins toxiques et biodégradables (COMNINELLIS et al., 1992). Par exemple, à l’anode les phénols sont oxydés en acide maléique, les cyanures en cyanates, les sulfures en sulfates, etc. (CALVIN et POON, 1997; COMNINELLIS, 1992). Il est important de souligner que ces électrodes dites à forte surtension d’oxygène permettent d’oxyder des composés non oxydables chimiquement ou difficilement oxydables (PULGARIN et al., 1994). Des molécules réputées non oxydables ou difficilement oxydables chimiquement comme l’acide éthylènediaminetétraacétique (EDTA) ou le chrome (III) peuvent être oxydées par voie électrochimique directe (PILLAUD, 1987). Plusieurs auteurs (BOCKRIS et al., 1990; COMNINELLIS, 1992) ont étudié les mécanismes d’oxydation électrochimique des molécules organiques. Par exemple, l’oxydation du phénol s’effectue généralement en deux étapes :
1re étape : Décharge anodique de l’eau : production de radicaux sur l’électrode M
2e étape : Oxydation de produits organiques R par les radicaux OHo
où (M) représente un site actif de l’électrode sur lequel les radicaux peuvent s’adsorber et (RO) le produit organique oxydé. Par ailleurs, l’électrolyse directe peut également avoir un effet sur les bactéries. Une étude réalisée par GARNERONE (1979) indiquait qu’une électrolyse de suspensions concentrées d’Escherichia coli, sur des électrodes poreuses en charbon actif aggloméré, entraînait une désactivation des bactéries. Le champ électrique peut aussi avoir une action bactéricide. En effet, de part et d’autre des membranes bactériennes, il existe un champ électrique qui, vu la faible épaisseur de ces membranes, est de l’ordre de quelques centaines de volts par centimètre. Lorsqu’une solution contaminée est soumise à un champ électrique supérieur au champ existant au niveau de la membrane bactérienne, un effet bactéricide est observé (VERNHES et al., 1999).
7.2 Oxydation anodique indirecte
Dans certains cas, l’électrolyse peut être inefficace ou lente. Il est alors nécessaire de générer électrochimiquement un oxydant qui réagira par la suite en solution avec les polluants. L’action indirecte peut être obtenue par oxydation d’halogénures (Cl‑, Br‑ ou I‑) ou par réduction de l’oxygène dissout, lesquelles réactions permettent de générer des oxydants bactéricides tels que HClO, HBrO, IOH, I3- ou H2O2 (CANIZARES et al., 2002, 2005).
L’oxydation des halogénures, notamment les chlorures et les bromures, est souvent utilisée en désinfection des eaux. L’efficacité bactéricide de ces oxydants dépend de leur capacité à traverser la membrane bactérienne. Les membranes bactériennes seraient plus perméables aux formes moléculaires qu’aux formes ioniques (BUNCE, 1990). Ainsi, les molécules HClO et HBrO seraient plus bactéricides que les composés ioniques ClO- et BrO-. Des études réalisées sur l’effet combiné du courant électrique et d’oxydants chimiques montrent que les bactéries, initialement soumises à un courant électrique, deviennent plus sensibles aux oxydants chimiques et sont plus rapidement éliminées (DROGUI et al., 2001). Ces oxydants bactéricides (notamment HClO), permettent également d’oxyder un grand nombre de molécules organiques (phénol, acides humiques, etc.) et inorganiques (H2S, CN-, Cr(VI), etc.) contenues dans les eaux usées.
7.3 Application et intégration des systèmes d’oxydation électrochimique dans les usines de traitement des eaux et d’effluents
L’aspect non polluant de l’électricité, la facilité d’automatisation et la réduction du volume d’équipements dans les procédés de traitement des eaux usées sont autant de paramètres qui concourent au développement industriel des techniques électrolytiques, et ce, comparativement aux procédés conventionnels (chimique ou biologique). Plusieurs systèmes électrolytiques ont été développés à l’échelle industrielle pour le traitement d’effluents contaminés par divers polluants organiques (phénol, acides humiques, acides fulviques, etc.) et inorganiques (métaux lourds, phosphates, sulfures, fluorures, etc.). En revanche, très peu de procédés électrochimiques ont été développés à l’échelle industrielle pour le traitement des eaux usées municipales ou industrielles. Vers les années 1970, HILLIS (1970) étudiait déjà la possibilité d’intégrer des systèmes électrolytiques dans les usines d’épuration des eaux usées. Dans son étude, il décrivait un système électrolytique installé dans une usine d’épuration municipale située à proximité d’une zone côtière sur l’île de Guernesey. Ce système permettait d’assurer une désinfection et une désodorisation des eaux usées avant leur rejet dans le milieu récepteur (en mer). Les plages étaient ainsi décontaminées et les baigneurs étaient mieux protégés contre toute contamination bactérienne. La chaîne de traitement était constituée d’une cellule électrolytique (anode : Ti/Pt; cathode : Fe) dans laquelle l’eau de mer était électrolysée en vue de la production d’hypochlorite de sodium (NaClO). Des doses variant de 1 à 2 % NaClO (par rapport au débit d’écoulement des eaux à traiter) étaient injectées dans la conduite des eaux brutes, lesquelles avaient au préalable subi un traitement de désintégration visant à réduire en dessous de 3 mm le diamètre des solides totaux. Par la suite, le mélange d’eaux usées et de NaOCl était acheminé dans deux réacteurs de type cuve agitée ayant chacun un temps de rétention de 30 min. Une fois traité, l’effluent est rejeté en mer avec une concentration résiduelle de NaOCl de 2 mg•L‑1. Cette concentration résiduelle permettait d’assurer un effet rémanent. La consommation énergétique totale du système (incluant la consommation énergétique des pompes, du système de désintégration et de la cellule électrolytique) était estimée à 0.33 kWh•m‑3. Cette dépense énergétique est en grande partie attribuée au fonctionnement de la cellule électrolytique.
Une étude similaire a été également menée par MENDIA (1982) dans une usine d’épuration des eaux usées municipales située dans une région côtière de la ville de Naples en Italie. L’objectif de ces travaux était non seulement de réduire le volume d’installation des équipements de l’usine d’épuration, mais également de minimiser les coûts d’opération, et ce, en intégrant un système électrolytique dans la chaîne de traitement. Les eaux étaient initialement soumises à un dégrillage puis un dessablage avant d’être acheminées par la suite dans un réservoir ou elles étaient mélangées avec de l’eau de mer (ou eau salée) dans des proportions adéquates (ratio eau de mer/eau usée : 1:3; 1:4). Le mélange est par la suite acheminé dans une cellule électrolytique (anode : graphite; cathode : fer) ayant un temps de rétention de quelques secondes (5-10 sec). Une fois électrolysé, l’effluent était acheminé dans un bassin de floculation/clarification à l’intérieur duquel la désinfection et la sédimentation (des particules en suspension) s’effectuaient simultanément. Le temps de rétention du bassin de clarification/floculation était compris entre 60 à 110 min. Le surnageant était rejeté en mer alors que la fraction solide (boues) était séchée (lits de séchage) avant toute disposition finale à cause de sa teneur élevée en chlore. Il est important de souligner que le procédé complet était effectué pendant une période de temps inférieure à la moitié du temps requis lors du traitement conventionnel physico-chimique des eaux usées. Par conséquent, les unités de traitement pouvaient être plus petites et moins encombrantes. Les conditions opératoires ainsi que quelques résultats expérimentaux obtenus par Mendia sont résumés au tableau 7. Ces résultats étaient obtenus à partir de deux séries d’essais effectués à l’aide de six cellules électrolytiques montées en parallèle, lesquelles étaient connectées électriquement en série et fonctionnaient chacune avec des temps de rétentions respectifs de 5 et 10 sec. L’examen de ce tableau indique que la consommation énergétique est réduite de moitié, la DBO5 et la turbidité sont respectivement réduites de 3 % et 21 % lorsque le temps de rétention imposé est de 5 sec comparativement à 10 sec. Des concentrations de 2,56 et 7,36 mg•L‑1 de chlore résiduel étaient enregistrées à la sortie du bassin de clarification/floculation pour des temps de rétention respectifs de 5 et 10 sec. En revanche, après 24 h (après traitement), la concentration en chlore résiduel dans l’effluent est nulle lorsqu’un temps de rétention de 5 sec était imposé dans la cellule électrolytique alors qu’une concentration résiduelle de 5,2 mg•L‑1 était enregistrée pour un temps de rétention de 10 sec.
Tableau 7
Résultats expérimentaux des essais préindustriels effectués par MENDIA (1982).
Preindustrial experimental data recorded by MENDIA (1982).
Paramètres |
Essais |
|
|---|---|---|
1re série |
2e série |
|
Conditions opératoires | ||
Intensité de courant (A) |
90 |
90 |
Densité de courant anodique (A•dm-2) |
2,5 |
2,5 |
Différence de potentiel (par cellule) (V) |
4,4 |
4,0 |
Différence de potentiel totale (V) |
26,4 |
12 |
Consommation énergétique (kWh•m-3 d’eau usée) |
0,66 |
0,30 |
Débit d’alimentation (m3•h‑1) |
4,8 |
4,8 |
Temps de rétention dans la cellule électrolytique (sec) |
10 |
5 |
Temps de rétention dans le clarificateur /floculateur (min) |
105 |
105 |
Ratio : eau de mer/eau usée |
1 : 3 |
1 : 3 |
Caractéristiques de l’effluent traité | ||
Cl2 résiduel après électrolyse (mg•L‑1) |
10,2 |
5,1 |
Cl2 résiduel après clarification/floculation (mg•L‑1) |
7,36 |
2,56 |
Cl2 résiduel après 24 h (mg•L‑1) |
5,2 |
0 |
Efficacité épuratoire | ||
Réduction des MES (%) |
100 |
100 |
Réduction de la turbidité (%) |
76 |
56 |
Réduction de la DBO5 (%) |
62 |
59 |
FENG et al. (2003) ont développé un système de traitement électrochimique à l’échelle pilote pour le traitement d’eaux usées résiduaires. Ce système électrochimique combinant à la fois l’électrooxydation et l’électrocoagulation a montré son efficacité pour le traitement d’eaux usées domestiques et d’eaux usées issues de bassins contenant des algues. La chaîne de traitement était constituée d’une cellule d’électrooxydation comprenant une anode catalytique en Ti/RuO2-TiO2 et une cathode en acier inoxydable. La cellule fonctionnait avec une densité de courant de 3 mA•cm‑2, un temps de séjour de 15 min et un débit d’alimentation de 0,3 m3•h‑1. Ce procédé permettait d’enregistrer un abattement de la DCO de 86,3 % dans le cas d’effluents domestiques et de 92,4 % pour les eaux contenant des algues.
YEON et al. (2007) ont pour leur part montré l’efficacité d’un procédé pilote combinant à la fois électrooxydation et electrodialyse pour le traitement d’eaux usées issues d’une usine nucléaire située en Corée. L’unité d’électrooxidation employée dans ce procédé est constituée de 23 électrodes en Ti/RuO2-SnO2-IrO2 placées en mode bipolaires et permettait un abattement moyen de la DCO de 89 % pour un temps de résidence de 25 min, un débit de 6,0 L•h‑1 et une densité de courant de 70 mA•cm‑2.
En somme, les traitements électrochimiques constituent de nouvelles méthodes de traitement des eaux et des effluents visant, d’une part, à réduire la quantité de réactifs (diminution du volume de boues générées) et, d’autre part, à réduire le volume d’équipements (diminution du temps de rétention dans le réacteur). Plusieurs processus physiques et chimiques peuvent simultanément intervenir lors du traitement électrolytique d’un effluent (oxydation anodique, réduction cathodique, adsorption, flottation, etc.), lesquels contribuent dans la plupart des cas à une meilleure efficacité épuratoire comparativement aux techniques conventionnelles chimiques ou biologiques couramment utilisées.
8. Comparaison quantitative chiffrée des performances des POA
Les applications des POA pour la dégradation des polluants organiques réfractaires sont nombreuses. Sont proposées, dans le tableau 8, quelques applications réalisées à l’échelle de banc d’essai en laboratoire pour l’oxydation des composés organiques rentrant pour la plupart dans la catégorie des composés perturbateurs endocriniens. Au nombre de ces composés se trouvent des hormones et dérivés hormonaux (ex. oestradiol) les pesticides (ex. atrazine), les colorants (ex. Orange G), les analgésiques (ex. paracétamol), les composés phénoliques (ex. bisphénol A, phénol, chlorophénol), etc. Un taux de dégradation allant jusqu’à 100 % peut être obtenu dépendamment du POA appliqué, du type de composé organique et des conditions de traitement imposées. Parmi les composés organiques testés, le phénol reste de loin le composé qui a été le plus étudié par plusieurs auteurs afin de mettre en évidence la performance des POA pour la dégradation des polluants réfractaires. Par exemple, DE et al. (2006), en appliquant le procédé Fenton (H2O2/Fe2+) à un effluent synthétique contenant du phénol (C0 = 1,0 mM) obtiennent une dégradation totale de ce composé après une heure de traitement en présence de 44 mM de H2O2. Par comparaison, l’application du procédé de peroxonation (ratio molaire H2O2/O3 = 2,34 : 1) mettant également en jeu le peroxyde d’hydrogène (6,2 ‑ 6,3 mM H2O2) a permis d’obtenir un taux de dégradation de 80 % d’une solution aqueuse de phénol (C0 = 1,0 mM) après un temps de traitement de 80 min (IJEPLAAR et al., 2000). Les essais réalisés par MA et al. (2005) et IJEPLAAR et al. (2000) peuvent être comparés à ceux effectués par ESPLUGAS (2000) lors de la dégradation du phénol (C0 = 1,0 mM) par photolyse du peroxyde d’hydrogène (7,4 mM) en présence d’une source d’irradiation UV couplage H2O2/UV). Un taux de dégradation de 91 % a été enregistré au bout de 30 min de traitement. La comparaison de ces trois expérimentations (H2O2/Fe2+, H2O2/O3 et H2O2/UV) réalisées sur le même type de substrat (phénol et C0 = 1,0 mM), montre que le réactif de Fenton reste de loin le procédé qui requiert le plus de H2O2 (6 à 19 fois plus de H2O2 comparativement à l’application des procédés H2O2/O3 et H2O2/UV) pour atteindre une dégradation maximale du phénol.
Tableau 8
Comparaison des différents POAs.
Comparison of different advanced oxidation processes (AOPs).
PAOs |
Type d’eau |
Type de polluants |
Conditions opératoires |
Résultats et commentaires |
Références |
|---|---|---|---|---|---|
O3/H2O2 |
Eau souterraine |
Atrazine et herbicides |
pH 7,8 ratio = 3,3 g•g‑1 H2O2 = 8,8 mg•L‑1 |
80 % d’élimination d’atrazine et herbicides; 99 % d’élimination d’atrazine |
IJEPLAAR et al. (2000) |
Eau potable |
Acide clofibrique Ibuprofen dichlorfenac |
O3 = 1,0 mg•L‑1; TRH = 10 min; C0 = 2μg•L‑1 (polluant) Ratio molaire O3/H2O2 = 2 :1 |
Enlèvement au bout de 10 min : 10 % acide clofibrique, 30 % ibuprofen et 100 % dichorfenac. |
ZWIENER et FRIMMEL (2000). |
|
Effluent déionisée |
Colorant (Red -RR2) |
C0= 40 mg•L‑1, débit d’ozone = 500 mL•min‑1, [H2O2]=1 000 mg•L‑1. T = 25 °C. |
Après 30 min : 27 % de décoloration à pH 4, 49 % à pH 7 et 57 % à pH 10. |
WU et al. (2008) |
|
Solution aqueuse |
Phénol |
C0=1 mmol•L‑1 ; H2O2 = 6,2-6,8 mM ; (H2O2 / O3 = 2,34 : 1) V = 2,5 L, débit recirculation = 100 L•h‑1. |
Après 80 min, 80 % de dégradation à pH entre 3,4 et 5,0; 93 % à pH 6,8 et 93 % à pH 9,3. |
Esplugas (2002). |
|
H2O2/UV |
Eau distillée |
Paracétamol |
Lampe basse pression (17 W, 254 nm); H2O2= 5,0 et 20 mmol•L‑1; pH 2,0-7,0 |
Élimination complète du paracétamol avec une minéralisation comprise entre 21 % et 40 % en utilisant respectivement 5,0 et 20 mmol•L‑1 de H2O2 |
ANDREOZZI et al (2003) |
Solution aqueuse |
Acide clofibrique |
Lampe basse pression (17 W, 254 nm); C0= 1,0 mmol•L‑1; H2O2 = 1,0 mol•L‑1; pH 5; TRH = 60 min |
Une élimination quasi totale de l’acide clofibrique; légère minéralisation |
ANDREOZZI et al (2003) |
|
Eau distillée |
Carbamazépine |
Lampe basse pression (254 nm) C0=20 mol•L‑1; H2O2 = 5 mmol•L‑1; pH = 5 |
100 % d’enlèvement de carbamazépine après 4 min de traitement et 35 % d’enlèvement de TOC. |
Vogna et al. (2004). |
|
Solution aqueuse |
Colorant (Red -RR198) |
C0=20 mg•L‑1 et H2O2 =2 mM, pH 7. Source irradiation : 15 W, lampe UVC |
100 % de décoloration après 50 min |
WU (2007). |
|
Solution aqueuse |
Phénol |
C0=1 mmol•L‑1, H2O2=7,4 mM ; V = 1,5 L; débit recirculation = 60 L•h‑1; pH = 2,3-3,2 |
91 % après 30 min contre 24 % sans irradiation. |
Esplugas (2002). |
|
TiO2/UV |
Solution aqueuse |
17β oestradiol |
150 W lamp; C0 = 0,005-3,0 μmol•L‑1; |
50 % de 17β oestradiol était éliminé en 40 min et 98 % d’élimination en 3,5 h |
Cleman et al. (2000) |
Eau distillée |
Bisphénol A (BPA) |
200 W Hg–Xe lampe; C0 = 175 mol•L‑1; I=10 mW•cm-2; TiO2 en suspension. |
100% minéralisation du bisphénol A après 20 h; activité ostrogénique réduite à 10 % après 6 h de traitement. |
Ohko et al. (2001) |
|
Effluent synthétique |
Colorants : orange G, acide Orange 7, acide black 1, tartrazine, Jaune acide |
TiO2 =3 g•L‑1; irradiation par lampe à mercure de 50 W |
75 % d’enlèvement entre 60 et 240 min. Le facteur déterminant pour la dégradation est l’adsorption du colorant sur TiO2. |
Tanaka et al. (2000) |
|
Effluent synthétique |
Phénol |
TiO2 =1 g•L‑1; irradiation par lampe à mercure haute pression de 400 W, volume du réacteur = 0,8 L. |
Dégradation >90 % pour C0<0,13 mM. pH optimal = 7,4. La pré-humidification de TiO2 améliore la photodégradation. |
Chiou et al. (2008). |
|
H2O2/Fe2+ |
Eaux souterraines |
Atrazine |
pH 5,5; H2O2 = 10 mg•L‑1; Fe2+ = 5,1 mg•L‑1 |
75 % d’enlèvement d’atrazine. |
IJEPLAAR et al. (2000) |
Effluent synthétique |
p-Chlorophénol |
pH étudié entre 1 et 5 Fe2+ = 0,1-1 mM ; H2O2 = 3-12 mM. |
pH optimum =3 ; Fe2+ est le paramètre limitant pour l’efficacité de dégradation. |
Kwon et al. 1999. |
|
Effluent synthétique |
Phénol ; ortho et para-chlorophénol, |
Phénol =1,06 mol•m-3 ; V = 300 mL. Chlorophenol= 0,78 mol•m-3 FeSO4= 0,079 mol•m-3 H2O2= 44,1mol•m-3 (H2O2/Fe2+ = 550:1) |
100 % de dégradation après 1 h H2O2 et Fe2+ ont des effets significatifs sur la dégradation de contaminants. |
DEet al. 2006. |
|
Solution aqueuse |
Benzène sulfonate de sodium |
Fe3+=2.10-4 M ; H2O2 =4.10-3 M , C0=2.10-4 M |
100 % de dégradation après 60 min. 40 % enlèvement TOC après 4 h. Amélioration de l’efficacité de dégradation en présence d’irradiation |
MA et al. (2005) |
|
Électrooxydation (EO) |
Solution aqueuse |
Phénol |
I = 57 mA•cm-2 pH=2 , conc. initiale en phénol = 21 mmol•L‑1 |
90 % d’enlèvement TOC après le passage de 50 Ah•dm-3 avec SnO2 comme anode et 38 % avec Pt (anode). |
Comninellis 1992 |
Solution aqueuse |
Bisphenol A (BPA) |
Anode : BDD ; C0 = 20 mg•L‑1 ; Na2SO4 = 0,1M ; pH = 6 |
100% de dégradation à I=35,7mA•cm-2 après 4 h et 50 % à I = 14,3 mA•cm-2 |
Murugananthan et al. (2007) |
|
Solution aqueuse |
17β estradiol (E2) |
Anode BDD ; C0 = 500 μg•L‑1 Na2SO4 = 0,1M |
100 % de dégradation au bout de 40 min d’électrolyse à I = 24 mA•cm-2 |
Murugananthanet al. (2006) |
|
Solution aqueuse |
Trichloroethylene (TCE) |
C0 = 5 mg•L‑1 |
Après 10 h de traitement, 8 %TCE transformé en chlorométhane à 5 V, 36 % à 10 V et 52 % à 20 V. |
Al-Abed et al. 2001. |
|
Sonochimie |
Solution aqueuse |
Phénol |
Ultrasons émis par disque piézoélectrique pour 500 kHz et par sonde en Ti pour 20 kHz. |
Au bout de 150 min : 70 % d’enlèvement à 500 kHz et moins de 10 % à 20 kHz. |
Entezari et al. (2003) |
Solution aqueuse |
Colorant (Red -RR 198) |
C0 = 20 mg•L‑1, Source d’irradiation : 40 kHz et 400 W. |
Après 120 min seulement 16 % de décoloration à pH7 et 31 % à pH10. pas d’amélioration en présence d’H2O2. |
WU (2007). |
|
Solution aqueuse |
Nitrobenzene (NB) p-nitrophenol (pNP) |
C0(NB) = 100 ppm ; C0(pNP) = 10 ppm, V = 150 mL; T=25°C source d’irradiation : 20 kHz, 100 W |
Après 90 min de sonication : seulement 30 % de NB dégradé et moins de 5 % pour le pNP. |
Sivasanka et al. (2008). |
|
Eau dé-ionisée (1), eau de mer (2), lessivage eau urbaine (3), eau usée (4), solution aqueuse NaCl 3,5% (5). |
Triclosan |
C0 = 5μg•L‑1 ; V = 200 mL Source d’irradiation : 80 kHz, 135 W. |
~100 % de triclosan dégradé après 120 min excepté pour l’effluent d’eau usée (60 % après 180 min). Vitesse de dégradation différente suivant le type de matrice : (2)>(5)>(3)>(1)>(4). |
Sanchez-Pradoet al. (2008). |
|
Solution aqueuse |
Hydrazine |
C0 = 0,1 et C0 = 1 mmol•L‑1 ; V=100 mL Source d’irradiation : 200 kHz, 200 W |
60 min sonication : 40 % dégradation pour C0 = 0,1 mmol•L‑1 à pH 8,4 et 20 % pour C0 = 1 mmol•L‑1 à pH 8,7. |
Nakui et al. (2007). |
Par ailleurs, il peut être également intéressant de comparer la dégradation du phénol à partir des POA (procédés d’oxydation sono-chimique et électrochimique) utilisant directement l’électricité comme source d’énergie dans le processus d’oxydation de polluants. COMNINELLIS (1992) a étudié plusieurs types d’électrodes anodiques en titane recouvert d’un dépôt catalytique (Pt, RuO2, IrO2, PbO2 et SnO2) pour le traitement électrochimique d’une solution aqueuse (volume utile 0,15 dm3) contenant du phénol (C0 = 21 mM). La meilleure efficacité épuratoire était obtenue en utilisant le titane recouvert d’oxyde d’étain (Ti/SnO2), laquelle électrode permettait d’obtenir un taux d’enlèvement de 90 % de carbone organique total (COT) en appliquant une quantité de charge de 50 A•h‑1•dm‑3 et une tension moyenne de 4,2 V. Ce taux de minéralisation du phénol permet d’estimer la consommation spécifique d’énergie électrique consommée, laquelle se situe à 117,6 kWh•kg‑1 de phénol éliminé. En comparaison, une valeur de 8 101 kWh•kg‑1 est estimée lors des essais de dégradation sonochimique du phénol (C0 = 0,67 mM) réalisés par ENTERAZZI et al. (2003) à l’aide d’une cellule de sonification comprenant un disque piézoélectrique et fonctionnant sous une fréquence de 20 kHz et une puissance de 50 W. Un taux de dégradation du phénol de 70 % a été enregistré au bout de 150 min de traitement. Cette consommation d’énergie très élevée enregistrée dans le cas du traitement sonochimique (soit 69 fois plus élevée que celle enregistrée dans le cas de l’électrooxydation) est l’une des raisons principales qui limitent le développement industriel de ce type de procédé. Dans le but de minimiser cette dépense énergétique, les ultrasons ne sont pas employés seuls. Ils sont souvent utilisés en synergie avec d’autres procédés (notamment, les procédés chimiques et électrochimiques) (voir section 6.2). De nombreuses études réalisées à l’échelle laboratoire ont clairement prouvé l’efficacité des POA pour le traitement de divers effluents (effluents synthétiques, eaux usées, effluents industriels, etc.). Cependant, le développement de ces procédés à grande échelle (dans les filières de potabilisation ou de traitement des eaux usées) reste encore limité en raison des coûts d’investissement et des coûts opératoires associés, lesquels étant nettement supérieurs aux procédés conventionnels. Les domaines majeurs pour la recherche et le développement futurs des POA sont indiqués dans la section suivante.
9. Développements actuels et futurs des POA
Les procédés d’oxydation avancée (POA) sont souvent employés pour la dégradation des polluants organiques réputés non oxydables ou difficilement oxydables par des procédés chimiques ou biologiques conventionnels. Le tableau 9 indique certains avantages et inconvénients des POA lors du traitement des eaux et des effluents. Certains procédés comme O3/UV et TiO2/UV sont souvent limités par des phénomènes de transfert de matière, alors que d’autres comme les procédés d’oxydation sonochimique sont très coûteux d’un point de vue de la consommation énergétique. Les radicaux hydroxyles (OHo) générés lors des POA sont des espèces hautement actives susceptibles de réagir avec la quasi-totalité des molécules organiques. Ces espèces radicalaires réagissent avec les doubles liaisons carbone/carbone et attaquent les noyaux aromatiques, composants majeurs des composés organiques réfractaires. Il y a trois domaines majeurs pour la recherche et le développement futurs des POA : i) les POA doivent davantage s’adapter et s’intégrer dans les filières existantes de traitement des eaux et des effluents. Le couplage O3/H2O2 est un bel exemple qui peut être avantageusement appliqué dans le cas de la modernisation des usines possédant déjà un système d’ozonation; ii) en plus des installations moins encombrantes (compactes) et faciles d’automatisation (réduction des coûts d’opération), les recherches doivent davantage êtres menées pour réduire les concentrations de réactifs de manière à rendre les POA plus compétitifs que les autres procédés (chimiques, physiques ou biologiques) et iii) les traitements combinés impliquant les POA doivent être développés pour améliorer l’efficacité des traitements biologiques, physiques et chimiques vis-à-vis des polluants (organiques, inorganiques et microbiens) réfractaires et émergents (GEBHARDT et SCHROEDER, 2007; LAFI et AL-QODAH, 2006).
Tableau 9
Performance des POAs pour la dégradation des polluants organiques réfractaires.
Performance of advanced oxidation processes (AOPs) for the degradation of refractory organic compounds.
Techniques |
Avantages |
Inconvénients |
|---|---|---|
O3/UV |
Oxydant puissant; Production relativement simple de radicaux libres; Formation de H2O2, ce qui permet d’assurer un effet rémanent lors de la désinfection des eaux. |
Traitement onéreux; réaction limitée par le transfert de masse; Efficacité et durée de vie des lampes UV limitées; Minéralisation souvent incomplète; sous-produits inconnus. |
H2O2/Fe2+ (Fenton) |
Oxydant puissant; décoloration rapide de la solution; minéralisation des colorants organiques. |
Requiert un apport constant en H2O2; régénération limitée du catalyseur; Transport, stockage et manipulation de H2O2. |
H2O2/UV |
Source importante et directe de radicaux hydroxyles; 100% de rendement quantique de la photolyse. |
Transport, stockage et manipulation de H2O2; Efficacité et durée de vie des lampes UV limitée; faible coefficient d’absorption de H2O2; production de sous-produits inconnus. |
TiO2/UV |
Installation simple; dégradation efficace des composés organiques. |
Limitation par le transfert de masse; nécessité de récupération du catalyseur après réaction; efficacité et durée de vie limitée des lampes UV. |
Électro-oxydation |
Élimination efficace des composés organiques; pas d’ajout supplémentaire de réactifs chimiques; action directe et indirecte du courant électrique. |
Efficacité de courant diminue au cours du traitement; réaction parasite (formation de O2 moléculaire). |
Sonochimie |
Excellent transfert de masse; plusieurs modes dégradation : pyrolyse, cisaillement, attaque par les radicaux OH0. |
Procédé coûteux; très sensible aux caractéristiques de l’effluent. |
10. Conclusions
La qualité des eaux entrant dans les usines de production d’eau potable et dans les stations d’épuration des eaux usées est de plus en plus affectée par la contamination de composés organiques réfractaires issus de différentes industries, chimiques, pharmaceutiques, agroalimentaires, cosmétiques, pétrolières, etc. Au nombre des ces polluants couramment décelés dans les rejets industriels et urbains, se trouvent des hormones et dérivés hormonaux, des composés phénoliques, des antibiotiques, des produits phytosanitaires, des organochlorés, des produits cosmétiques, etc., lesquels composés sont à l’origine de nombreuses perturbations de la faune aquatique et constituent un risque pour la santé humaine. Les normes en matière de rejet de ces effluents vers les milieux naturels devenant de plus en plus sévères, les POA se révèlent de bons candidats pour résoudre cette problématique. Malgré cela, le développement de ces procédés dans les filières de traitement des eaux reste limité. La principale raison est que les coûts d’investissement, ainsi que les coûts opératoires sont nettement plus élevés que ceux relevant des techniques conventionnelles employées dans le traitement des eaux. En vue de favoriser le développement à grande échelle des POA, cette synthèse sur les applications des POA suggère les solution suivantes : 1) adapter davantage les POA aux usines existantes de traitement des eaux afin d’accroître leur efficacité épuratoire (vis-à-vis des polluants émergents et réfractaires) sans procéder nécessairement à la construction majeure de nouvelles infrastructures; 2) développer des stratégies d’opération afin de réduire autant que possible les quantités de réactifs tout en assurant une efficacité épuratoire permettant d’atteindre les normes de rejet ou de potabilisation qui évoluent vers une sévérité croissante; 3) les POA peuvent avantageusement être couplés à des procédés biologiques lors du traitement des effluents contenant des composés organiques réfractaires. Ce type de couplage permet à la fois de tirer profit des avantages des POA (temps de rétention court) et de la biodégradation (faibles coûts d’opération). Utilisés en synergie avec les procédés biologiques, les POA peuvent êtres employés pour transformer les composés non biodégradables en produits biodégradables, ou encore comme traitement final pour l’oxydation complète des composés organiques en dioxyde de carbone.
L’efficacité des POA vis-à-vis de la dégradation des polluants résulte de la génération in situ d’espèces radicalaires très réactives, principalement les radicaux hydroxyles. Ils ont une durée de vie extrêmement courte qui n’excède pas le dixième de microseconde. Ils peuvent instantanément oxyder la plupart des composés organiques par des réactions d’addition et des réactions d’abstraction d’hydrogène. Cependant, certains composés organiques, comme ceux saturés en chlore (ex. les organochlorés) sont difficilement dégradés par les radicaux hydroxyles. En effet, leur structure très équilibrée au niveau de la répartition des charges leur confère une résistance à toute attaque nucléophile, car les sites électrophiles sont inexistants. Par ailleurs, l’efficacité de la plupart des POA exige un faible TAC (titre alcalimétrie complet) car les ions carbonates et bicarbonates piègent les radicaux hydroxyles.
Les POA agissent en général suivant deux modes d’action, soit par action directe ou par action indirecte. Par exemple, dans les procédés d’oxydation mixte (O3/UV, H2O2/UV, O3/H2O2), les oxydants chimiques tels que O3 et/ou H2O2 peuvent réagir directement sur le polluant cible (action directe) avec une cinétique relativement faible comparativement à l’action indirecte obtenue par génération de radicaux hydroxyles.
Parties annexes
Références bibliographiques
- AGUIAR A. (1992). Dégradation de quelques pesticides en milieux aqueux lors de l’oxydation du Fe(II) en Fe(III) par le peroxyde d’hydrogène. Thèse de Doctorat, Université de Poitiers, Poitiers, France, 158 p.
- AGUIAR A., F. CARBONNIÈRE, H. PAILLARD et B. LEGUBE (1993). Oxydation des pesticides et coagulation des substances humiques par le peroxyde d’hydrogène à faibles doses et le fer ferreux. Water Supply, 11, 129-138.
- AKMEHMET B.I. et M. OTKER (2004). Pre-treatment of antibiotic formulation wastewater by O3, O3/H2O2, and O3/UV processes. Turkish J. Eng. Environ. Sci., 28, 325‑331.
- AL-ABED S.R., J.-L. CHEN, V.R. KUKAINIS, S. LIANG et K.M. KORAN (2001). The effect of voltage on electrochemical degradation of trichloroethylene. Dans : Symposia Papers Presented before the Division Environmental Chemistry American Chemical Society, San Diego, CA.
- AMAT A.M., A. ARQUES, M.A. MIRANDA, R. VINCENTE et S. SEGUI (2007). Degradation of two commercial anionic surfactants by means of ozone and/or UV irradiation. Environ. Eng. Sci., 24, 790-794.
- ANDREOZZI R., V. CAPRIO, R. MAROTTA et D.VOGNA (2003). Paracetamol oxidation from aqueous solutions by means of ozonation and H2O2/UV system. Water Res., 37, 993–1004.
- ANDREOZZI R., V. CAPRIO, R. MAROTTA et A. RADOVNIKOVIC (2003). Ozonation and H2O2/UV treatment of clofibric acid in water: a kinetic investigation. J. Hazard. Mater., 103, 233–246.
- AURIOL M., Y. FILALI-MEKNASSI et R.D. TYAGI (2007). Présence et devenir des hormones stéroïdiennes dans les stations de traitement des eaux usées. Rev. Sci. Eau, 20, 89-108.
- AZABOU S., W. NAJJAR, A. GARGOUBI, A. GHORBEL et S. SAYADI (2007). Catalytic wet peroxide photo-oxidation of phenolic olive oil mill wastewater contaminants. Part II. Degradation and detoxification of low-molecular mass phenolic compounds in model and real effluent. Appl. Cat. B: Environ., 77, 166-174.
- BARB G., J. BAXENDALE et R. HARGRAVE (1951). Reaction of ferrous and ferric ions with hydrogen peroxide. Trans. Faraday Soc., 47, 462-500, 591-615.
- BARBENI M., C. MINERO et L. PELLIZZETTI (1987). Chemical degradation of chlorophenols with Fenton’s reagent. Chemosphere, 16, 2225-2232.
- BERGER J.A. et A.G. MARR (1960). Sonic disruption of spores of Bacillus cereus. J. Gen. Microbiol., 221, 147-157.
- BERLAN J., F. TRABELSI, H. DELMAS, A. WILHELM et M. PETRIGNANI (1994). Oxidative degradation of phenol in aqueous media using ultrasound. Ultrason. Sonochem., 1, S97-S102.
- BHOWMICK M. et M.J. SEMMENS (1994). Ultraviolet photo-oxidation for the destruction of VOCs in air. Water Res., 28, 2407-2415.
- BIELSKI B.H.J., D.E. CABELLI, R.J. ARUDI et A.B. ROSS (1985). Reactivity of HO2/O2- radical in aqueous solution. J. Phys. Ref. Data, 14, 1041-1100.
- BOCKRIS J., L. KABA et G. HITCHENS (1990). Electrochemical incineration of wastes. J. Electrochem. Soc., 137 1341-1345.
- BRILLAS E. et J. CASADO (2002). Aniline degradation by electro-Fenton and proxi-coagulation processes using a flow reactor wastewater treatment. Chemosphere, 47, 241‑248.
- BRILLAS E., J.C. CALPE et J. CASADO (1999). Mineralization of 2,4-D by advanced electrochemical oxidation processes. Water Res., 34, 2253-2262.
- BRILLAS E., R.M. BASTIDA, E.F. LLOSA et J. CASADO (1995). Electrochemical destruction of aniline and 4-chloroaniline for wastewater treatment using a carbon-PTFE O2-Fed cathode. Electrochem. Sci. Technol., 142, 1733-1741.
- BUNCE N.J. (1990). Environmental Chemistry. Wuerz Publishing, Winnipeg, Manitoba, Canada, 376 p.
- BUXTON G.U., C.L. GREENSTOCK, W.C HELMAN et A.B. ROSS (1988). Critical review of rate constant for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (HO/O-) in aqueous solution. J. Phys. Chem. Ref. Data, 17, 513-759.
- CALVIN P. et C. POON (1997). Electroflotation for groundwater decontamination. J. Hazard. Mater., 55, 159‑170.
- CANIZARES P., C. MARTINEZ, M. DIAZ, J. GARCIA-GOMEZ et M. A. RODRIGO (2002). Electrochemical oxidation of aqueous phenol wastes using active and nonactive electrodes. J. Electrochem. Soc., 149, 118-124.
- CANIZARES P., J. LOBATO, R. PAZ, M.A. RODRIGO, C.J. SAEZ et M.A. RODRIGO (2005). Electrochemical oxidation of phenolic wastes with boron-doped diamond anodes. Water Res., 39, 2687-2703.
- CHROMOSTAT N., J. DE LAAT, M. DORÉ, H. SUTY et M. POUILLOT (1993). Étude de la dégradation de triazine par O3/H2O2 et O3. Cinétique et sous-produits de dégradation. Water Supply, 11, 149-157.
- CHIOU C.H., C.Y. WU et R.-S. JUANG (2008). Photocatalytic degradation of phenol and m-nitrophenol using irradiated TiO2 in aqueous solutions. Separ. Purif. Technol., 62, 559–564.
- COLEMAN H.M., B.R. EGGINS, J.A. BYRNE, F.L. PALMER et E. KING (2000). Photocatalytic degradation of 17-[beta]-oestradiol on immibilised TiO2. Appl. Catal. B Environ, 24, L1-5.
- COLOBERT L., B. MONTAGNON. et C. NOFRE (1962). Effet microbicide sur Escherichia coli de systèmes chimiques générateurs du radical libre hydroxyle. Mécanisme de l’activité bactéricide du peroxyde d’hydrogène et de l’acide ascorbique. Ann. Inst. Pasteur, 102, 278-291.
- COMNINELLIS C. (1992). Electrochemical treatment of wastewater containing phenol. Trans. Inst. Chem. Eng., 70, 219-224.
- COMNINELLIS C., P. SEIGNEZ, P. PÉRINGER et E. PLATTNER (1992). Dégradation des polluants organiques industriels : traitement électrochimique, biologique et leur couplage. Swiss Chem., 14, 25-30.
- CRISSOT F. (1996). Oxydation catalytique de composés organiques aqueux par le peroxyde d’hydrogène en phase hétérogène. Thèse de Doctorat, Université de Poitiers, École Supérieure d’Ingénieurs de Poitiers, Poitier, France, 152 p.
- DE A.K., B.K. DUTTA et S. BHATTACHARJEE (2006). Reaction kinetics for the degradation of phenol and chlorinated phenols using Fenton’s reagent. Environ. Progress, 25, 64-71.
- DENG Y. et J.D. ENGLEHARDT (2006). Treatment of landfill leachate by Fenton process. Water Res., 40, 3683‑3694.
- DORÉ M. (1989). Chimie des oxydants et traitement des eaux. Tec. Doc. Lavoisier (Éditeur), Paris, France, 505 p.
- DROGUI P., S. ELMALEH, M. RUMEAU, C. BERNARD et A. RAMBAUD (2001). Oxidising and disinfecting by hydrogen peroxide produced in a two-electrode cell. Water Res., 35, 3235-3241.
- DROGUI P., J.F. BLAIS et G. MERCIER (2007). Review of electrochemical technologies for environmental applications. Recent Patent Eng., 1, 257-272.
- DUGUET J.P. et L. CARPENTIER (1989). Technique de désinfection alternative. Lyonnaise des Eaux, Veille Technologique, Novembre, 19 p.
- DURAN MORENO A., B.A. FONTANA-URIBE et R.M. RAMIREZ ZAMORA (2004). Electro-Fenton as a feasible advanced treatment process to produce reclaimed water. Water Sci. Technol., 50, 83-90.
- ECKENFELDER W., A.R. BOWERS et J.A. ROTH (1992). Chemical oxidation: technologies for the nineties. Dans: Chemical Oxidation: Technology for the Nineties, Proceedings of the first international symposium, Vanderbilt University, Nashville, Tennessee, 20 au 22 février 1991, Technomic Pub. Co., Lancaster, PA.
- EL-DEIN A.M. (2002). Oxidation kinetics of the azo dye reactive black 5 using H2O2/UV and aerobic biological treatment. Verfahrenstechnik, 762, 1-134.
- ENTEZARI M.H., C. PETRIER, P. DEVIDAL (2003). Sonochemical degradation of phenol in water: a comparison of classical equipment with a new cylindrical reactor. Ultrason. Sonochem., 10, 103-108.
- ESPLUGAS S., J. GIMENEZ, S. CONTRERAS, E. PASCUAL et M. RODRIGUEZ (2002). Comparison of different advanced oxidation processes for phenol degradation. Water Res., 36, 1034‑1042.
- FENG C., N. SUGIURA, S. SHIMADA et T. MAEKAWA (2003). Development of high performance electrochemical wastewater treatment system. J. Hazard. Mater., B103, 65‑78.
- FLOTRON V., C. DELTEIL, A. BERMOND et V. CAMEL (2003). Remediation of matrices contaminated by polycyclic aromatic hydrocarbon: use of Fenton’s reagent. Polycyclic Aromatic Compounds, 23, 353-376.
- GALEY C. et D. PASLAWSKI (1993). Élimination des micropolluants par l’ozone couplé avec le peroxyde d’hydrogène dans le traitement de potabilisation des eaux. L’Eau, L’Industrie, Les Nuisances, 161, 46-49.
- GANDINI D., C. COMNINELLIS, N.B. Tahar et A. Savall (1998). Électrodépollution : Traitement électrochimique des eaux résiduaires chargées en matières organiques toxiques. Actualité Chimique, 10, 68-73.
- GARNERONE A. (1979). Contribution à l’étude des mécanismes d’action des agents antiseptiques dans les eaux : Application à la désinfection électrochimique indirecte. Thèse de Doctorat, Institut National Polytechnique de Grenoble, France, 165 p.
- GEBHARDT W. et H.F. SCHROEDER (2007). Liquid chromatography-(tandem) mass spectrometry for the follow-up of the elimination of persistent pharmaceuticals during wastewater treatment applying biological wastewater treatment and advanced oxidation. J. Chromatography, 1160, 34-43.
- GOEL R.K., J.R.V. FLORA et J. FERRY (2003). Mechanisms for naphthalene removal during electrolytic aeration. Water Res., 37, 891-901.
- GOGATE P.R. et A.B. PANDIT, (2000a). Engineering design methods for cavitational reactors I: sonochemical reactors. AIChE J., 46, 372.
- GOGATE P.R. et A.B. PANDIT (2001). Hydrodynamic cavitation reactors: A state of the art review. Rev. Chem. Eng., 17,1-85.
- GOGATE P.R. et A.B. PANDIT (2004). A review of imperative technologies for wastewater treatment I: oxidation technologies at ambient conditions. Adv. Environ. Res., 8, 501-551.
- GONCHARUK V.V., N.G. POTAPCHENKO, O.S. SAVLUK, V.N. KOSINOVA et A.N. SOVA (2003). Study of various conditions for O3/UV disinfection of water. Khimiya i Tecknologiya Vody, 25, 487-496.
- GÖZMEN B., M.A. OTURAN, N. OTURAN et O. ERBATUR (2003). Indirect electrochemical treatment of biphenol A in water via electrochemical generated Fenton’s reagent. Environ. Sci. Technol., 37, 3716-3723.
- GUIVARCH Z.E. (2004). Traitement des polluants organiques en milieux aqueux par procédé électrochimique d’oxydation avancée «Électro-Fenton». Application à la minéralisation des colorants synthétiques. Thèse de Doctorat, Université de Marne-la-Vallée, Institut Francilien des Géosciences, Marne-la-Vallée, France, 235 p.
- HAAG W. et C.C.D. YAO (1992). Rate constant for reaction of hydroxyl radicals with several drinking water contaminants. Environ. Sci. Technol., 26, 1005-1013.
- HABER P. et J. WEISS (1934). The catalytic decomposition of hydrogen by iron salts. Proc. Royal Soc., A147, 332-351.
- HELMY S.M., S. EL RAFIE et M.Y. GHALY (2003). Bioremediation post-photo-oxidation and coagulation for black liquor effluent treatment. Desalination, 158, 331‑339.
- HERNANDEZ R., M. ZAPPI, J. COLLUCI et R. JONES (2002). Comparing the performance of various advanced oxidation process for treatment of acetone contaminated water. J. Hazard. Mater., 92, 33-50.
- HILLIS M.R. (1970). Treatment of effluents by electrolytic methods in Britain. Water. Pollut. Control, 108, 22-27.
- HSING H.J., P.C. CHIANG, E.E. CHANG et M.Y. CHEN (2007). The decolorization and mineralization of acid orange 6 azo dye in aqueous solution by advanced oxidation processes: A comparative study. J. Hazard. Mater., 141, 8-16.
- HUA I. et M.R. HOFFMANN (1997). Optimization of ultrasonic irradiation as an advanced oxidation technology. Environ. Sci. Technol., 31, 2237-2243.
- IJEPLAAR G.F., R.T.MEIJERS et R. HOPMAN (2000). Oxidation of herbicides in groundwater by the Fenton process: a realistic alternative for O3/H2O2 treatment? Ozone Sci. Eng., 22, 607‑616.
- IKEHATA K et M.G. EL-DIN (2006). Aqueous pesticide degradation by hydrogen peroxide/ultraviolet irradiation and Fenton-type advanced oxidation processes: a review. J. Environ. Eng. Sci., 5, 81-135.
- IKEHATA K, N.J. NAGHASHKAR et M.G. EL-DIN (2006). Degradation of aqueous pharmaceuticals by ozonation and advanced oxidation processes: a review. Ozone Sci. Eng., 28, 353-414.
- IRMAK S., O. ERBATUR et A. AKGERMAN (2005). Degradation of 17β-estradiol and bisphenol A in aqueous medium by using ozone and ozone/UV techniques. J. Hazard. Mater., 126, 54-62.
- JÜRGENS M.D., K.I.E. HOLTHAUS, A.C. JOHNSON, J.J.L. SMITH, M. HETHERIDGE et R.J. WILLIAMS (2002). The potential for estradiol and ethinylestradiol degradation in English rivers. Environ. Toxicol. Chem., 21, 480-488.
- KANG S.F., C.H. LIAO et H.P. HUNG (1999). Peroxidation treatment of dye manufacturing wastewaters in the presence of ultraviolet light and ferrous ions. J. Hazard. Mater.B., 65, 317-333.
- KANG S.F., C.H. LIAO et S.T. PO (2000). Decolorization of textile wastewater by photo-Fenton oxidation technology. Chemosphere, 41, 1287-1294.
- KARAM L.R., D.S. BERGTOLDS et M.G. SIMIC (1991). Biomarkers of HO radicals damage in-vivo. Free Radical. Res., 12/13, 11-16.
- KARPEL VEL LEITNER N. et M. DORÉ (1997). Mécanisme d’action des radicaux OH° sur les acides glycolique, glyoxylique, acétique et oxalique en solution aqueuse : Incidence sur la consommation de peroxyde d’hydrogène dans les systèmes H2O2/UV et O3/H2O2. Water Res., 31, 1383-1397.
- KUO W.G. (1992). Decolorizing dye wastewater with Fenton’s reagent. Water Res., 26, 881-886.
- KUO W.G. (1992). Decolorizing dye wastewater with Fenton’s reagent. Water Res., 26, 881-886.
- KULKARNI A.G., R. TANDON et R.M. MATHUR (2006). Some chemical aspects of color removal from effluents of paper industry. Ippta J., 18, 55-61.
- KWON B.G., D.S. LEE, N. KANG et J. YOON (1999). Characteristics of p-chlorophenol oxidation by Fenton’s reagent. Water Res., 33, 2110.
- LAFI W.K. et Z. AL-QODAH (2006). Combined advanced oxidation and biological treatment processes for the removal of pesticides from aqueous solutions. J. Hazard. Mater., 137, 489-497.
- LAU T.K., W. CHU et N.GRAHAM (2007). Degradation of the endocrine disruptor carbofuran by UV, O3 and O3/UV. Water Sci. Technol., 55, 275-280.
- LI Z.M., P.J. SHEA et S.D. COMFORT (1997a). Fenton oxidation of 2,4,6-trinitrotoluene in contaminated soil slurries. Environ. Eng. Sci., 14, 55-66.
- LI Z.M., S.D. COMFORT et P.J. SHEA (1997b). Destruction of 2,4,6-trinitrotoluene by Fenton oxidation. J. Environ. Qual., 26, 480-487.
- LIN S.H. et C.C. LO (1997). Fenton process for treatment of desizing wastewater. Water Res., 31, 2050-2056.
- LORIMER, J.P., T.J. MASON, (1987). Sonochemistry. Part1 – The physical aspects. Chem. Soc. Rev., 16, 239-274.
- MA J., W. SONG, C. CHEN, W. MA, J. ZHAO et Y. TANG (2005). Fenton degradation of organic compounds promoted by dyes under visible irradiation. Environ. Sci. Technol., 39, 5810-5815.
- MASON T.J. (1990). A survey of commercially available sources of ultrasound suitable for sonochemistry. Sonochemistry – Uses of Ultrasound in Chemistry. Royal Society of Chemistry, Cambridge UK, 158 p.
- MAESTRE P. (1991). Radical hydroxyle et métaux redox : application à la toxicité des quinones. Thèse de Doctorat, Université de Toulouse, Toulouse, France, 237 p.
- MARTINEZ-HUITLE C.A et S. FERRO (2006). Electrochemical oxidation of organic pollutants for the wastewater treatment: direct and indirect processes. Chem. Soc. Rev., 35, 1324-1340.
- MATSUE T., M. FUJIHIRA et T. OSA (1981). Oxidation of alkylbenzenes by electrogenerated hydroxyl radicals. J. Electrochem. Soc., 128, 2565-2569.
- MENDIA L. (1982). Electrochemical processes for wastewater treatment. Water Sci. Technol., 14, 331-344.
- MERTZ J. et W. WATERS (1949). Some oxidation involving the free hydroxyl radical. J. Chem. Soc., 15, 515-525.
- MILLET M. (1992a). L’oxygène et les radicaux libres (1re partie). Bios, 23, 67-70.
- MILLET M. (1992b). L’oxygène et les radicaux libres (2e partie). Bios, 23, 45-50.
- MILLS A. et S. LE HUNTE (1997). An overview of semiconductor photocatalysis. J. Photochem. Photobiol. A: Chem., 108, 1-35.
- MIRANDA M.A., A.M. AMAT et A. ARQUES (2001). Abatement of the major contaminants present in olive oil industry wastewaters by different oxidation methods: Ozone and/or UV radiation versus solar light. Water Sci. Technol., 44, 325-330.
- MOISEEV A., H. SCHROEDER, M. KOTSARIDOU-NAGEL, S.U. GEISSEN et A. VOGELPOHL (2004). Photocatalytic polishing of paper-mill effluents. Water Sci. Technol., 49, 325-330.
- MURUGANANTHAN M., S.YOSHIHARA, T.RAKUMA, N. UEHARA et T. SHIRAKASSHI (2007). Electrochemical degradation of 17β-Estradiol (E2) at boron-doped diamond (Si/BDD) thin electrode. Electrochim. Acta, 52, 3242-3249.
- MURUGANANTHAN M., S.YOSHIHARA, T.RAKUMA, T. SHIRAKASSHI (2008). Mineralization of bisphenol A (BPA) by anodic oxidation with boron-doped diamond (BDD) electrode. J. Hazard. Mater., 154, 213-220.
- NAFFRECHOUX E., E COMBET, B. FANGET et C. PETRIER (2003). Reduction of chloroform formation potential of humic acid by sonolys and ultraviolet irradiation. Water Res., 37, 1948-1952.
- NAJJAR W., S. AZABOU, S. SAYADI et A. GHORBEL (2007). Catalytic wet peroxide photo-oxidation of phenolic olive oil mill wastewater contaminants. Appl. Cat. B Environ., 74, 11-18.
- NAKUI H., K. OKITSU, Y. MAEDA, R. NISHIMURA (2007). The effect of pH on sonochemical degradation of hydrazine. Ultrason. Sonochem., 14, 627–632.
- OHKO Y., I. ANDO, C. NIWA, T. TATSUMA, T. YAMAMURA, T. NAKASHIMA, Y. KUBOTA et A. FUJISHIMA (2001). Degradation of bisphenol A in water by TiO2, photocatalyst. Environ. Sci. Technol., 35, 2365‑2368.
- OTURAN M.A., J.J. AARON, N. OTURAN et J. PINSON (1999). Degradation of chlorophenoxy acid herbicides in aqueous media, using a novel electrochemical method. Pestic. Sci., 55, 558-562.
- OTURAN M.A., N. OTURAN, C. LAHITTE et S. TRÉVIN (2001). Production of hydroxyl radical by electrochemically assisted Fenton’s reagent application to the mineralization of an organic micropollutant: pentachlorophenol. J. Electroanal. Chem., 507, 96-102.
- OTURAN N.M et J. PINSON (1992). Polyhydroxylation of salicylic acid by electrochemically generated HOo radicals. New J. Chem., 16, 705-710.
- PAILLARD H. (1994). Étude de la minéralisation de la matière organique dissoute en milieu aqueux dilué par ozonation, oxydation avancée O3/H2O2 et ozonation catalytique hétérogène. Thèse de Doctorat de l’université de Poitiers, Poitiers, France, 224 p.
- PAILLARD H., R. BRUNET et M. DORÉ (1988). Conditions optimales d’application du système oxydant ozone-peroxyde d’hydrogène. Water Res., 22, 91-103.
- PANIZZA M., M. ZOLEZZI et C. NICOLELLA (2006). Biological and electrochemical oxidation of naphthalenesulfonates. J. Chem. Technol. Biotechnol., 81, 225-232.
- PARSONS S. (2004). Advanced oxidation processes for water and wastewater treatment. IWA Publishing, Alliance House, Londres, Angleterre, 356 p.
- PRAKASH N.S., E.T. PUTTAIAH, B.R. KIRAN, B.K. HARISH et K.M. MAHADEVAN (2007). Photo oxidation of textile industrial effluent in the presence of semiconductor particles by solar exposure. Res. J. Chem. Environ., 11, 73-77.
- PETRIER C., Y. JIANG, A. FRANCONY et M.F. LAMY (1999). Aromatics and chloroaromatics sonochemical degradation. Yields and byproducts. Ham. Ber. Siedlungswasserwirtschaft (Ultrasound in Environmental Engineering), 25, 23-37.
- PIGNATELLO J.J. (1992). Dark and photoassisted Fe3+‑catalysed degradation of chlorophenoxy herbicides by hydrogen peroxide. Environ. Sci. Technol., 26, 944-951.
- PILLAUD J.L. (1987). Application de l’oxydation anodique au traitement de certains effluents industriels. Thèse de Doctorat, Université de Montpellier II, Montpellier, France, 202 p.
- POTTER F.J. et J.A. ROTH (1993). Oxidation of chlorinated phenols using Fenton’s reagent. Hazard. Waste Hazard. Mater.,10, 157-170.
- PULGARIN C, N. ADLER, P. PERINGER et C. COMNINELLIS (1994). Electrochemical detoxification of a 1,4-benzoquinone solution in wastewater treatment. Water Res., 28, 887-893.
- RAJESHWAR K. et J.G. IBANEZ (1997). Environmental electrochemistry. Fundamentals and applications in pollution abatement. Academic Press, Londres, Angleterre, 776 p.
- RIEZ P., D. BERDAHL et C.L. CHRISTMAN (1985). Free radical generation by ultrasound in aquaous and nonaquaous solutions. Environ. Health Perspect., 64, 233‑252.
- SANCHEZ-PRADO L., R. BARRO, C. GARCIA-JARES, M. LLOMPART, M. LORES, C. PETRAKIS, N. KALOGERAKIS, D. MANTZAVINOS et E. PSILLAKIS (2008). Sonochemical degradation of triclosan in water and wastewater. Ultrason. Sonochem., 15, 689–694.
- SAULEDA R. et E. BRILLAS (2001). Mineralization of aniline and 4-chlorophenol in acidic solution by ozonation with Fe2+ and UVA light. Appl. Cat. B. Environ., 29, 135-145.
- SCHALLER V. (1996). Oxydation électrochimique du phénol sur des électrodes du type métal support/dépôt conducteur. Thèse de Doctorat, École Polytechnique Fédérale de Lausanne, Lausanne, Suisse, 250 p.
- SERVOS M.R., D.T. BENNIE, B.K. BURNISON, A. JURKOVIC, R. MCINNIS, T. NEHELI, A. SCHNELL, S.A. SMYTH et T.A. TERNES (2005). Distribution of estrogens, 17β-estradiol and estrone, in Canadian municipal wastewater treatment plants. Sci. Tot. Environ., 336, 155-170.
- SHANER E.O. (1964). Augmentation of bactericidal action of germicides with ultrasound. J. Acoust. Soc. Am., 36, 2238‑2239.
- SHU H.Y. et M.C. CHANG (2004). Decolorization effects of six azo dyes by O3, UV/O3 and UV/H2O2 processes. Dyes Pigm., 65, 25-31.
- SIVASANKAR T. et V.S. MOHOLKAR (2008). Physical features of sonochemical degradation of nitroaromatic pollutants. Chemosphere, 72, 795‑1806.
- SNYDER S.A., P. WERSTERHOFF, Y. YOON et D.L. SEDLAK (2003a). Pharmaceuticals, personal care products, and endocrine disruptors in water: Implication for water industry. Environ. Eng. Sci., 20, 449-469.
- SNYDER S.A., S.A. ADHAM, A.M. REDDING, F.S. CANNON, J. DECAROLIS, J. OPPENHEIMER, E.C. WERT et Y. YOON (2003b). Role of membrane and activated carbon in the removal of endocrine disruptors and pharmaceuticals. Desalination, 202, 156-181.
- STAEHELIN S. et J. HOIGNE (1985). Decomposition of ozone in water rate of initiation by hydroxide and hydrogen peroxide. Environ. Sci. Technol., 16, 676-681.
- STRIEBIG B.A., J.M. SCHNEIDER, T.A. SPAEDER, M.R. MALLERY, R.J. HEINSOHN et F.S. CANNON (1996). Analysis of advanced oxidation process in a hybrid air pollution control system. Dans: Chemical oxidation: Technology for the nineties, Proceedings of the International Chemical Oxidation Association Symposium, 14 au 17 avril, Nashville, TN.
- STRIOLO P. (1992). Oxydation d’effluents organiques aqueux par le peroxyde d’hydrogène à haute température : Procédé W.P.O. Thèse de Doctorat, Institut National des Sciences Appliquées de Toulouse, Toulouse, France.
- SUN Y. et J.J. PIGNATELLO (1993). Photochemical reactions involved in the total mineralization of 2,4-D by Fe3+/H2O2/UV. Environ. Sci. Technol., 27, 304-310.
- SUTY H., C. DE TRAVERSAY et M. COSTE (2003). Application of advanced oxidation processes: Present and future. Dans: Proceedings of the 3rd Conference on Oxidation Technologies for Water and Wastewater Treatment, 18 au 22 mai, Goslar, Allemagne, pp. 8.
- SZPYRKOWICZ L., S.N. KAUL et R.N. NETI (2005). Tannery wastewater treatment by electro-oxidation coupled with a biological process. J. Appl. Electrochem., 35, 381-390.
- TANAKA S., Y. NAKATA, T. KIMURA, M. YUSTIAWATI, M. KAWASAKI et H. KURAMITZ (2002). Electrochemical decomposition of bisphenol A using Pt/Ti and SnO2/Ti anodes. J. Appl. Electrochem., 32, 197-210.
- TANAKA, K., K. PADERMPOLE, T. HISANAGA. (2000). Photocatalytic degradation of commercial azo dyes. Water Res., 34, 327.
- TANG W.Z. et S. TASSOS (1997). Oxidation kinetics and mechanisms of trihalomethanes by Fenton’s reagent. Water Res., 31, 1117-1125.
- TONG S.P., Y.Q. CHU., C.A. MA et W.P. LIU (2005). The degradation of acetic acid and nitrobenzene in water by O3/UV. Zhongguo Huanjing Kexue, 25, 366-369.
- TRABELSI F., H. AÏT-LYAZIDI, B. RATSIMBA, A.M. WILHEM, H. DELMAS, P-L. FABRE et J. BERLAN (1996). Oxidation of phenol in wastewater by sonoelectrochemistry. Chem. Eng. Sci., 51, 1857-1865.
- USEPA (1997). Special report on environmental endocrine disruption: An effect assessment and analysis. U.S. Environmental Protection Agency, EPA/630/R-96/012, Washington, D.C.
- VAN CRAEYNEST K., H. VAN LANGENHOVE et R.M. STUETZ (2004). AOPs for VOCs and odour treatment. Dans : Advanced Oxidation Processes for Water and Wastewater Treatment. PARSONS S. (Éditeur), IWA Publishing, Alliance House, Londres, Angleterre, 356 p.
- VERNHES M.C., A. BENICHOU et P. PERNIN (1999). Éradication des amibes libres par l’utilisation de champs électriques pulsés. Dans : Journée d’Électrochimie, 1 au 4 juin 1999, Toulouse, France.
- VERSTRAETEN I.M., T. HEBERER, J.R. VOGEL, T. SPETH, S. ZUEHLKE et U. DUENNBIER (2003). Ocurrence of endocrine-disrupting and other wastewater compounds during water treatment with case studies from Lincol, Nebraska and Berlin, Germany. Pract. Period. Hazard. Toxic. Rad. Waste Manag. ASCE, 7, 253-263.
- VOGNA D., R. MAROTTA, R. ANDREOZZI, A. NAPOLITANO et M. D’ISCHIA (2004). Kinetic and chemical assessment of the UV/H2O2 treatment of antiepileptic drug carbamazepine, Chemosphere, 54, 497‑505.
- WANG T.H, S.F. KANG et Y.H. LIN (1999). Comparison among Fenton-related processes to remove 2,4-dinitrophenol. J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., A34, 1267-1281.
- WATTS R.J., A.P. JONES, P.H. CHEN et A. KENNY (1997). Mineral-catalyzed Fenton-like oxidation of sorbed chlorobenzenes. Water Environ. Res., 69, 269-275.
- WATTS R.J., M.D. UDELL et P.A. RAUCH (1990). Treatment of pentachlorophenol-contaminated soils using Fenton’s reagent. Hazard. Waste Hazard. Mater., 7, 335‑345.
- WU C.H. et C.L. CHANG (2006). Decolorization of reactive red 2 by advanced oxidation processses: comparative studies of homogeneous and heterogeneous systems. J. Hazard. Mater., 128, 265-272.
- WU C.H., H.Y. NG (2008). Degradation of C.I. Reactive Red 2 (RR2) using ozone-based systems: Comparisons of decolorization efficiency and power consumption. J. Hazard. Mater. 152, 120–127.
- WU C.H. (2007). Sonocatalytic degradation of C.I. reactive red 198 in H2O2-based systems. React. Kinet. Catal. Lett., 92, 377-384.
- XIONG F. (1990). Contribution à l’étude d’ozonisation des acides fulviques aquatiques. Thèse de 3e cycle, Université de Poitiers, Poitiers, France.
- XU Y.L., D.J. ZHONG et J.P. JIA (2008). Electrochemical-assisted photodegradation of Allura Red and textile effluent using a half-exposed rotating TiO2/Ti disc electrode. J. Environ. Sci. Health Part A Toxic/Hazard. Subst. Environ. Eng., 43, 503-510.
- YEON K.H., J.H. SONG, J. SHIM, S.H. MOON, Y.U. JEONG, (2007). H.Y. Joo Integrating electrochemical processes with electrodialysis reversal and electro-oxidation to minimize COD and T-N at wastewater treatment facilities of power plants. Desalination, 202, 400‑410.
- YONAR T., G.K. YONAR, K. KESTIOGLU et N. AZBAR (2005). Decolorization of textile effluent using homogeneous photochemical oxidation processes. Color. Technol., 121, 258-264.
- YOSHIDA M., B.D. LEE et M. HOSOMI (2000). Decomposition of aqueous tetrachloroethylene by Fenton oxidation treatment. Water Sci. Technol., 42, 203-208.
- ZWIENER C. et F.H. FRIMMEL (2000). Oxidative treatment of pharmaceuticals in water. Water Res., 34, 1881‑1885.
Liste des figures
Figure 1
Différents procédés d’oxydation avancée.
Different advanced oxidation processes..
Figure 2
Oxydation du phénol par les radicaux hydroxyles (DORÉ, 1989).
Phenol oxidation by hydroxyl radicals (DORÉ, 1989).
Figure 3
Traitement d’effluent par le procédé Fenton (GOGATE et PANDIT, 2004)
Effluent treatment using the Fenton process (GOGATE and PANDIT, 2004)
Figure 4
Mécanisme d’activation du peroxyde d’hydrogène par l’ozone (DORÉ, 1989).
Activation mechanism of H2O2 by ozone, (DORÉ, 1989).
Figure 5
Photo-excitation d’un semi-conducteur
Photoexcitation of a semiconductor.
Liste des tableaux
Tableau 1
Comparaison des potentiels normaux d’oxydoréduction des principaux oxydants utilisés dans le domaine du traitement des eauxé
Comparison of the oxido-reduction potential of oxidizing agents used in water treatment..
Couple redox |
Réactions |
Potentiel, (V/ENH), 25oC |
Références |
|---|---|---|---|
Cl2/Cl- |
Cl2(g) + 2e → 2Cl− |
1,36 |
DORÉ (1989) |
Br2/Br- |
Br2(l) + 2e → 2Br− |
1,06 |
DORÉ (1989) |
I2/I- |
I2 + 2e → 2I− |
0,53 |
DORÉ (1989) |
OH°/H2O |
OH° + H+ + e → H2O |
2,81 |
Schaller (1996) |
O3/O2 |
O3 + 2H+ + 2e → O2 + H2O |
2,07 |
Guivarch (2004) |
H2O2/H2O |
H2O2 + 2H+ + 2e → 2H2O |
1,77 |
DORÉ (1989) |
MnO4-/Mn2+ |
MnO−4 + 8H+ + 5e → Mn2+ + 4H2O |
1,51 |
DORÉ (1989) |
HClO/Cl- |
HClO + H+ + 2e → Cl− + H2O |
1,49 |
DORÉ (1989) |
ClO2/ClO2- |
ClO2 + e → ClO−2 |
0,95 |
DORÉ (1989) |
S2O82-/SO42- |
S2O2−8 + 2e → 2SO2−4 |
2,05 |
DORÉ (1989) |
Tableau 2
Oxydabilité des composés organiques par les radicaux hydroxyles (MERTZ et WATERS, 1949).
Organic compound oxidation by hydroxyl radicals (MERTZ et WATERS, 1949)..
Composés oxydables par un mécanisme en chaîne |
|---|
Alcools primaires et secondaires : dérivés méthyl, éthyl, n-propyl, isopropyl |
Hydroxy-acides : acides glycollique, lactique, hydroxybutyrique, thioglycolique |
Éthers : diéthyl éther, dioxane, tétrahydrofuranne, tétrahydrofuranne, tétra hydropyranne |
Aldéhydes : formol, acétaldéhydes |
Acides aminés : glycine, alanine |
Composés oxydables par un mécanisme sans chaîne |
Alcools tertiaires : butanol, alcoolamylique, pinacol, phényldiméthylcarbinol |
Esters : acétate de méthyl, acétate d’éthyle, acétate d’isopropyle |
Acides carboxyliques : acide propionique, butyrique, valérique, succinique, adipique |
Amines : diéthylamines, triéthylamines, pyridine |
Glycols : éthylènes, butylène |
Composés non oxydables par les radicaux OH0 |
Acides carboxyliques : acétique, malonique, maléique, fumarique |
Cétones : acétone, méthyl-éthylène-cétone |
Amides : urée, acétamides |
Tableau 3
Comparaison des constantes de vitesse pour l’ozone et les radicaux hydroxyles lors de l’oxydation de quelques molécules organiques (PARSONS, 2004).
Comparison of kinetic rate constants for the production Comparison of kinetic rate constants for the production of ozone and hydroxyl radicals recorded while oxidizing some molecular organic compounds (PARSONS, 2004).
Composés organiques |
Constantes cinétiques (M‑1 sec‑1) |
|
|---|---|---|
O3 |
OHo |
|
Benzène |
2,0 |
7,8 x 109 |
Toluène |
14 |
7,8 x 109 |
Chlorobenzène |
0,75 |
4,0 x 109 |
Trichloroethylène |
17 |
4,0 x 109 |
Tétrachloroéthylène |
<0,10 |
1,7 x 109 |
n-Butanol |
0,60 |
4,6 x 109 |
t-Butanol |
0,03 |
0,4 x 109 |
Tableau 4
Constantes cinétiques de réaction des radicaux hydroxyles en milieu aqueux sur les composés aliphatiques.
Kinetic rate constants of hydroxyl radical production in aqueous media while oxidizing aliphatic compounds.
Composés aliphatiques |
Substituants |
KHO (x 10‑7 M‑1 sec‑1) |
Références |
|---|---|---|---|
Méthane |
‑H |
11 |
Buxton et al. (1988) |
Méthanol |
‑OH |
97 |
Buxton et al. (1988) |
Méthylamine |
‑NH2 |
420 |
Buxton et al. (1988) |
Ion méthyl ammonium |
‑NH3+ |
3,5 |
Buxton et al. (1988) |
Diméthyl éther |
‑OCH3 |
100 |
Buxton et al. (1988) |
Acétone |
‑COCH3 |
11 |
Buxton et al. (1988) |
Acétonitrile |
‑CN |
2,2 |
Buxton et al. (1988) |
Acétamide |
‑CONH2 |
19 |
Buxton et al. (1988) |
Acide acétique |
‑COOH |
1,6 |
Buxton et al. (1988) |
Acétaldéhyde |
‑COH |
73 |
Buxton et al. (1988) |
Dibromométhane |
‑Br2 |
9,0 |
Haag et Yao (1992) |
Trichlorométhane |
‑Cl3 |
5,4 |
Haag et Yao (1992) |
Tableau 5
Composés aromatiques monosubstitués (BUXTON et al., 1988) et polysubstitués (HAAG et YAO, 1992).
Monosubstituted (BUXTON et al., 1988) and polysubstituted (HAAG and YAO, 1992) aromatic compounds.
Composés aromatiques monosubstitués |
Substituants |
KHO (x 10‑9 M‑1 sec‑1) |
Composés aromatiques polysubstitués |
KHO (x 10‑9 M‑1 sec1) |
|---|---|---|---|---|
Benzène |
‑H |
7,8 |
Phtalates |
4,0 |
Phénol |
‑OH |
14 |
Trichlorobenzène |
4,0 |
Aniline |
‑NH2 |
15 |
Dichlorobenzène |
5,0 |
Anisole |
‑OCH3 |
5,4 |
BPCs |
6,0 |
Toluène |
‑CH3 |
3,0 |
HAPs |
1,0 |
Éthylbenzène |
‑CH2CH3 |
7,5 |
Lindane |
5,2 |
Acide benzoïque |
‑COOH |
4,3 |
Atrazine |
2,6 |
Ion benzoate |
‑COO- |
5,9 |
Simazine |
2,8 |
Benzaldéhyde |
‑CHO |
4,4 |
Alschlor |
4,0 |
Acétophénone |
‑C(O)CH3 |
5,9 |
Carbofurane |
7,0 |
Nitrobenzène |
‑NO2 |
3,9 |
Pentachlorophénol |
4,0 |
Benzamide |
‑CONH2 |
3,4 |
Dinoseb |
4,0 |
Chlorobenzène |
‑Cl |
5,5 |
- |
- |
Fluorobenzène |
‑F |
10 |
- |
- |
Tableau 6
Exemples de composés organiques pouvant être photominéralisés par le TiO2 (MILLS et LE HUNTE, 1997).
Examples of organic compounds that may be photomineralized by TiO2 (MILLS and LE HUNTE, 1997).
Classes |
Exemples |
|---|---|
Alcanes |
Méthane, isobutane, heptane, cyclohexane, paraffine |
Haloalcanes |
Mono-, di-, tri-, tétrachlorométhane, tribromoéthane |
Alcools |
Méthanol, iso-propyl alcool, glucose, sucrose |
Acides carboxyliques |
Formique, éthanoïque, diméthyléthanoique, oxalique |
Alcènes |
Propène, cyclohexène |
Haloalcènes |
Perchloroéthène, dichloroéthène, 1,1,2-trichloroéthène |
Aromatiques |
Benzène, naphtalène |
Haloaromatiques |
(Di)-chlorobenzène, bromobenzène |
Nitrohaloaromatiques |
3,4-dichloronitrobenzène |
Phénols |
Phénol, hydroquinone, catéchol, méthylcatéchol, crésol |
Halophénols |
Pentachlorophénol, fluorophénol, difluorophénol |
Acides carboxyliques aromatiques |
Benzoïque, aminobenzoïque, phtalique, salicylique, hydroxybenzoïque, chlorohydroxybenzoïque |
Polymères |
Polyéthylène, PVC |
Surfactants |
SDS, polyéthylène glycol, dodécyl benzène, sulfonate, triméthylphosphate, tétrabutyl ammonium phosphate |
Herbicides |
Méthyl viologen, atrazine, simazine, prometron, propetryne et bentazon |
Pesticides |
DDT, parathion, lindane |
Colorants |
Bleu de méthylène, rhodamine B, orange de méthyle, fluoresceine |
Tableau 7
Résultats expérimentaux des essais préindustriels effectués par MENDIA (1982).
Preindustrial experimental data recorded by MENDIA (1982).
Paramètres |
Essais |
|
|---|---|---|
1re série |
2e série |
|
Conditions opératoires | ||
Intensité de courant (A) |
90 |
90 |
Densité de courant anodique (A•dm-2) |
2,5 |
2,5 |
Différence de potentiel (par cellule) (V) |
4,4 |
4,0 |
Différence de potentiel totale (V) |
26,4 |
12 |
Consommation énergétique (kWh•m-3 d’eau usée) |
0,66 |
0,30 |
Débit d’alimentation (m3•h‑1) |
4,8 |
4,8 |
Temps de rétention dans la cellule électrolytique (sec) |
10 |
5 |
Temps de rétention dans le clarificateur /floculateur (min) |
105 |
105 |
Ratio : eau de mer/eau usée |
1 : 3 |
1 : 3 |
Caractéristiques de l’effluent traité | ||
Cl2 résiduel après électrolyse (mg•L‑1) |
10,2 |
5,1 |
Cl2 résiduel après clarification/floculation (mg•L‑1) |
7,36 |
2,56 |
Cl2 résiduel après 24 h (mg•L‑1) |
5,2 |
0 |
Efficacité épuratoire | ||
Réduction des MES (%) |
100 |
100 |
Réduction de la turbidité (%) |
76 |
56 |
Réduction de la DBO5 (%) |
62 |
59 |
Tableau 8
Comparaison des différents POAs.
Comparison of different advanced oxidation processes (AOPs).
PAOs |
Type d’eau |
Type de polluants |
Conditions opératoires |
Résultats et commentaires |
Références |
|---|---|---|---|---|---|
O3/H2O2 |
Eau souterraine |
Atrazine et herbicides |
pH 7,8 ratio = 3,3 g•g‑1 H2O2 = 8,8 mg•L‑1 |
80 % d’élimination d’atrazine et herbicides; 99 % d’élimination d’atrazine |
IJEPLAAR et al. (2000) |
Eau potable |
Acide clofibrique Ibuprofen dichlorfenac |
O3 = 1,0 mg•L‑1; TRH = 10 min; C0 = 2μg•L‑1 (polluant) Ratio molaire O3/H2O2 = 2 :1 |
Enlèvement au bout de 10 min : 10 % acide clofibrique, 30 % ibuprofen et 100 % dichorfenac. |
ZWIENER et FRIMMEL (2000). |
|
Effluent déionisée |
Colorant (Red -RR2) |
C0= 40 mg•L‑1, débit d’ozone = 500 mL•min‑1, [H2O2]=1 000 mg•L‑1. T = 25 °C. |
Après 30 min : 27 % de décoloration à pH 4, 49 % à pH 7 et 57 % à pH 10. |
WU et al. (2008) |
|
Solution aqueuse |
Phénol |
C0=1 mmol•L‑1 ; H2O2 = 6,2-6,8 mM ; (H2O2 / O3 = 2,34 : 1) V = 2,5 L, débit recirculation = 100 L•h‑1. |
Après 80 min, 80 % de dégradation à pH entre 3,4 et 5,0; 93 % à pH 6,8 et 93 % à pH 9,3. |
Esplugas (2002). |
|
H2O2/UV |
Eau distillée |
Paracétamol |
Lampe basse pression (17 W, 254 nm); H2O2= 5,0 et 20 mmol•L‑1; pH 2,0-7,0 |
Élimination complète du paracétamol avec une minéralisation comprise entre 21 % et 40 % en utilisant respectivement 5,0 et 20 mmol•L‑1 de H2O2 |
ANDREOZZI et al (2003) |
Solution aqueuse |
Acide clofibrique |
Lampe basse pression (17 W, 254 nm); C0= 1,0 mmol•L‑1; H2O2 = 1,0 mol•L‑1; pH 5; TRH = 60 min |
Une élimination quasi totale de l’acide clofibrique; légère minéralisation |
ANDREOZZI et al (2003) |
|
Eau distillée |
Carbamazépine |
Lampe basse pression (254 nm) C0=20 mol•L‑1; H2O2 = 5 mmol•L‑1; pH = 5 |
100 % d’enlèvement de carbamazépine après 4 min de traitement et 35 % d’enlèvement de TOC. |
Vogna et al. (2004). |
|
Solution aqueuse |
Colorant (Red -RR198) |
C0=20 mg•L‑1 et H2O2 =2 mM, pH 7. Source irradiation : 15 W, lampe UVC |
100 % de décoloration après 50 min |
WU (2007). |
|
Solution aqueuse |
Phénol |
C0=1 mmol•L‑1, H2O2=7,4 mM ; V = 1,5 L; débit recirculation = 60 L•h‑1; pH = 2,3-3,2 |
91 % après 30 min contre 24 % sans irradiation. |
Esplugas (2002). |
|
TiO2/UV |
Solution aqueuse |
17β oestradiol |
150 W lamp; C0 = 0,005-3,0 μmol•L‑1; |
50 % de 17β oestradiol était éliminé en 40 min et 98 % d’élimination en 3,5 h |
Cleman et al. (2000) |
Eau distillée |
Bisphénol A (BPA) |
200 W Hg–Xe lampe; C0 = 175 mol•L‑1; I=10 mW•cm-2; TiO2 en suspension. |
100% minéralisation du bisphénol A après 20 h; activité ostrogénique réduite à 10 % après 6 h de traitement. |
Ohko et al. (2001) |
|
Effluent synthétique |
Colorants : orange G, acide Orange 7, acide black 1, tartrazine, Jaune acide |
TiO2 =3 g•L‑1; irradiation par lampe à mercure de 50 W |
75 % d’enlèvement entre 60 et 240 min. Le facteur déterminant pour la dégradation est l’adsorption du colorant sur TiO2. |
Tanaka et al. (2000) |
|
Effluent synthétique |
Phénol |
TiO2 =1 g•L‑1; irradiation par lampe à mercure haute pression de 400 W, volume du réacteur = 0,8 L. |
Dégradation >90 % pour C0<0,13 mM. pH optimal = 7,4. La pré-humidification de TiO2 améliore la photodégradation. |
Chiou et al. (2008). |
|
H2O2/Fe2+ |
Eaux souterraines |
Atrazine |
pH 5,5; H2O2 = 10 mg•L‑1; Fe2+ = 5,1 mg•L‑1 |
75 % d’enlèvement d’atrazine. |
IJEPLAAR et al. (2000) |
Effluent synthétique |
p-Chlorophénol |
pH étudié entre 1 et 5 Fe2+ = 0,1-1 mM ; H2O2 = 3-12 mM. |
pH optimum =3 ; Fe2+ est le paramètre limitant pour l’efficacité de dégradation. |
Kwon et al. 1999. |
|
Effluent synthétique |
Phénol ; ortho et para-chlorophénol, |
Phénol =1,06 mol•m-3 ; V = 300 mL. Chlorophenol= 0,78 mol•m-3 FeSO4= 0,079 mol•m-3 H2O2= 44,1mol•m-3 (H2O2/Fe2+ = 550:1) |
100 % de dégradation après 1 h H2O2 et Fe2+ ont des effets significatifs sur la dégradation de contaminants. |
DEet al. 2006. |
|
Solution aqueuse |
Benzène sulfonate de sodium |
Fe3+=2.10-4 M ; H2O2 =4.10-3 M , C0=2.10-4 M |
100 % de dégradation après 60 min. 40 % enlèvement TOC après 4 h. Amélioration de l’efficacité de dégradation en présence d’irradiation |
MA et al. (2005) |
|
Électrooxydation (EO) |
Solution aqueuse |
Phénol |
I = 57 mA•cm-2 pH=2 , conc. initiale en phénol = 21 mmol•L‑1 |
90 % d’enlèvement TOC après le passage de 50 Ah•dm-3 avec SnO2 comme anode et 38 % avec Pt (anode). |
Comninellis 1992 |
Solution aqueuse |
Bisphenol A (BPA) |
Anode : BDD ; C0 = 20 mg•L‑1 ; Na2SO4 = 0,1M ; pH = 6 |
100% de dégradation à I=35,7mA•cm-2 après 4 h et 50 % à I = 14,3 mA•cm-2 |
Murugananthan et al. (2007) |
|
Solution aqueuse |
17β estradiol (E2) |
Anode BDD ; C0 = 500 μg•L‑1 Na2SO4 = 0,1M |
100 % de dégradation au bout de 40 min d’électrolyse à I = 24 mA•cm-2 |
Murugananthanet al. (2006) |
|
Solution aqueuse |
Trichloroethylene (TCE) |
C0 = 5 mg•L‑1 |
Après 10 h de traitement, 8 %TCE transformé en chlorométhane à 5 V, 36 % à 10 V et 52 % à 20 V. |
Al-Abed et al. 2001. |
|
Sonochimie |
Solution aqueuse |
Phénol |
Ultrasons émis par disque piézoélectrique pour 500 kHz et par sonde en Ti pour 20 kHz. |
Au bout de 150 min : 70 % d’enlèvement à 500 kHz et moins de 10 % à 20 kHz. |
Entezari et al. (2003) |
Solution aqueuse |
Colorant (Red -RR 198) |
C0 = 20 mg•L‑1, Source d’irradiation : 40 kHz et 400 W. |
Après 120 min seulement 16 % de décoloration à pH7 et 31 % à pH10. pas d’amélioration en présence d’H2O2. |
WU (2007). |
|
Solution aqueuse |
Nitrobenzene (NB) p-nitrophenol (pNP) |
C0(NB) = 100 ppm ; C0(pNP) = 10 ppm, V = 150 mL; T=25°C source d’irradiation : 20 kHz, 100 W |
Après 90 min de sonication : seulement 30 % de NB dégradé et moins de 5 % pour le pNP. |
Sivasanka et al. (2008). |
|
Eau dé-ionisée (1), eau de mer (2), lessivage eau urbaine (3), eau usée (4), solution aqueuse NaCl 3,5% (5). |
Triclosan |
C0 = 5μg•L‑1 ; V = 200 mL Source d’irradiation : 80 kHz, 135 W. |
~100 % de triclosan dégradé après 120 min excepté pour l’effluent d’eau usée (60 % après 180 min). Vitesse de dégradation différente suivant le type de matrice : (2)>(5)>(3)>(1)>(4). |
Sanchez-Pradoet al. (2008). |
|
Solution aqueuse |
Hydrazine |
C0 = 0,1 et C0 = 1 mmol•L‑1 ; V=100 mL Source d’irradiation : 200 kHz, 200 W |
60 min sonication : 40 % dégradation pour C0 = 0,1 mmol•L‑1 à pH 8,4 et 20 % pour C0 = 1 mmol•L‑1 à pH 8,7. |
Nakui et al. (2007). |
Tableau 9
Performance des POAs pour la dégradation des polluants organiques réfractaires.
Performance of advanced oxidation processes (AOPs) for the degradation of refractory organic compounds.
Techniques |
Avantages |
Inconvénients |
|---|---|---|
O3/UV |
Oxydant puissant; Production relativement simple de radicaux libres; Formation de H2O2, ce qui permet d’assurer un effet rémanent lors de la désinfection des eaux. |
Traitement onéreux; réaction limitée par le transfert de masse; Efficacité et durée de vie des lampes UV limitées; Minéralisation souvent incomplète; sous-produits inconnus. |
H2O2/Fe2+ (Fenton) |
Oxydant puissant; décoloration rapide de la solution; minéralisation des colorants organiques. |
Requiert un apport constant en H2O2; régénération limitée du catalyseur; Transport, stockage et manipulation de H2O2. |
H2O2/UV |
Source importante et directe de radicaux hydroxyles; 100% de rendement quantique de la photolyse. |
Transport, stockage et manipulation de H2O2; Efficacité et durée de vie des lampes UV limitée; faible coefficient d’absorption de H2O2; production de sous-produits inconnus. |
TiO2/UV |
Installation simple; dégradation efficace des composés organiques. |
Limitation par le transfert de masse; nécessité de récupération du catalyseur après réaction; efficacité et durée de vie limitée des lampes UV. |
Électro-oxydation |
Élimination efficace des composés organiques; pas d’ajout supplémentaire de réactifs chimiques; action directe et indirecte du courant électrique. |
Efficacité de courant diminue au cours du traitement; réaction parasite (formation de O2 moléculaire). |
Sonochimie |
Excellent transfert de masse; plusieurs modes dégradation : pyrolyse, cisaillement, attaque par les radicaux OH0. |
Procédé coûteux; très sensible aux caractéristiques de l’effluent. |