Résumés
Résumé
La phosphorylation d’un récepteur couplé aux protéines G (RCPG) par une kinase spécifique, nommée GRK (G protein-coupled receptor kinase), est une première étape qui participe, avec l’action des arrestines, à l’arrêt de la transmission du signal, au cours d’un processus appelé désensibilisation. Le dérèglement de ce mécanisme de protection cellulaire, mis en évidence dans différentes situations pathologiques, relève soit de mutations génétiques d’une GRK ou d’une arrestine, soit d’une variation de leur expression. Ce dérèglement a pour conséquence de modifier l’activité des RCPG qui interviennent dans de nombreuses fonctions vitales de l’organisme. Ainsi, dans la maladie d’Oguchi, la stimulation excessive de la rhodopsine par la lumière, due à la perte de fonction de la GRK1 ou de l’arrestine 1, conduit à des problèmes d’adaptation de la vision à l’obscurité. La mise au point d’un modèle de souris hypertendues, après transfection ciblée du gène de la GRK2 au niveau des vaisseaux, suggère fortement que l’augmentation de cette GRK participe, chez l’homme, au développement de l’hypertension associée à une baisse de l’effet vasodilatateur des récepteurs β-adrénergiques. L’idée de rétablir une activité RCPG normale en agissant sur ces mécanismes de désensibilisation a été couronnée de succès dans des modèles animaux de défaillance cardiaque chronique, et laisse supposer que la modulation de l’activité des GRK ou de la fonction des arrestines pourrait constituer une piste thérapeutique. Cependant, la réalisation d’essais chez l’homme devra encore attendre la découverte de molécules pharmacologiques efficaces et non toxiques.
Summary
Phosphorylation of the agonist-activated form of G-protein-coupled receptors (GPCRs) by a protein kinase from the G-protein-coupled receptor kinase (GRK) family initiates, with arrestin proteins, a negative feedback process known as desensitization. Because these receptors are involved in so many vital functions, it seems likely that disorders affecting GRK- or arrestin-mediated regulation of GPCRs would contribute to, if not engender, disease. Traditionally, it is believed that the desensitization process protects the cell against an overstimulation; however, in certain situations, this process is maladjusted and participes in disease progression. For example, in Oguchi disease, excessive rhodopsin stimulation due to a functional loss of GRK1 or arrestin 1 leads to light sensitization and stationary night blindness. Also, transgenic mice with vascular smooth muscle-targeted overexpression of GRK2 showed an elevated resting blood pressure, suggesting that increase in GRK2 level in humans is involved in hypertension associated with a decreased effect of β-adrenergic receptor-mediated vasorelaxation. The restoration of normal GPCR function in modulating the desensitization process has been successfully demonstrated in animal models of heart failure, which indicates that targeting GRKs or arrestins may open a novel therapeutic strategy in human diseases with GPCR dysregulation. However, the few effective pharmacological compounds in this domain currently preclude human clinical tests.
Corps de l’article
Après activation par un agoniste, les récepteurs couplés aux protéines G (RCPG) déclenchent un processus de rétrocontrôle négatif de la stimulation, connu sous le nom de désensibilisation : ce processus permet l’arrêt de la transmission du signal et évite les effets potentiellement dangereux d’une stimulation excessive de la cellule. Les mécanismes moléculaires responsables de la désensibilisation débutent, en général, par la phosphorylation du récepteur par une GRK (kinases des récepteurs couplés aux protéines G), suivie de la fixation d’une arrestine empêchant le couplage du récepteur à une nouvelle protéine G [1] (Figure 1). Les GRK sont également capables de phosphoryler des cibles différentes des RCPG, mais dont la signification physiologique est encore souvent à élucider.
Figure 1
Mécanismes membranaires de la désensibilisation du récepteur β2-adrénergique.
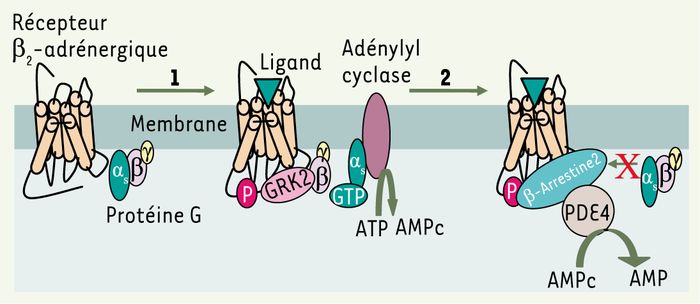
Ce récepteur, qui a fait l’objet d’études approfondies, est considéré comme un prototype dans la régulation de la transduction du signal couplée à l’activation de l’adénylyl cyclase. La fixation du ligand sur le récepteur β2-adrénergique provoque la synthèse de l’AMPc par l’adénylyl cyclase activée par la sous unité Gαs liée au GTP. Dans les instants qui suivent, la GRK2 est recrutée à la membrane plasmique, où elle fixe la sous-unité βγ et les phospholipides. La localisation de la GRK2 lui permet de phosphoryler des résidus sérine et thréonine de la partie carboxyterminale du récepteur (étape 1). Le récepteur β2-adrénergique phosphorylé par la GRK2 a une affinité accrue pour la β-arrestine 2, qui accentue le processus de désensibilisation en empêchant le couplage d’une nouvelle protéine Gs. De plus, la liaison de la phosphodiestérase 4 (PDE4) à la β-arrestine 2 contribue à la transformation de l’AMPc en AMP, et limite ainsi une stimulation excessive de la cellule par le second messager (étape 2).
Sept gènes codant pour des GRK (1 à 7) ont été clonés chez l’homme ; sur la base d’une homologie de leur séquence primaire, la famille des GRK a été subdivisée en trois sous-groupes : celui de la rhodopsine kinase (GRK1 et -7), celui des kinases spécifiques des récepteurs β-adrénergiques (GRK2 et -3) et celui de la GRK4 (GRK4, -5 et -6). La famille des arrestines, quant à elle, comprend quatre membres : deux arrestines visuelles, l’arrestine 1 et l’arrestine 4 (ou x-arrestine), et deux arrestines ayant une distribution plus ubiquitaire, l’arrestine 2 (ou β-arrestine 1) et l’arrestine 3 (ou β-arrestine 2). En plus de leur rôle dans la désensibilisation membranaire des RCPG, les β-arrestines peuvent entraîner l’internalisation des récepteurs, et déclencher d’autres voies de signalisation cytoplasmique en se comportant comme des protéines adaptatrices dans la formation d’échafaudage moléculaire [2] (Figure 2).
Le rôle clé du couple GRK/arrestine dans la désensibilisation des RCPG, ainsi que l’implication de ces récepteurs dans de nombreuses fonctions vitales de l’organisme suggèrent qu’un défaut de régulation des RCPG pourrait contribuer au développement de diverses pathologies. À la lumière des résultats récents concernant la physiopathologie des GRK et des arrestines [3], cet article fait le point sur leur implication dans certaines maladies humaines, et sur les conséquences thérapeutiques d’une modification de leur activité ou de leur expression chez l’animal.
Maladie d’Oguchi et régulation de la rhodopsine
La rhodopsine est un photorécepteur de la famille des RCPG, dont l’activation est régulée par la GRK1 et les arrestines visuelles. Des analyses génétiques et biochimiques ont révélé que les mutations du gène de la GRK1 ou de l’arrestine 1 sont associées à un dysfonctionnement du système visuel, connu sous le nom de maladie d’Oguchi. Cette maladie rare, transmise selon le mode autosomique récessif, est caractérisée par une coloration jaune ou grise du fond de l’oeil, et un temps d’adaptation prolongée à la vision nocturne. De plus, des études menées chez la souris déficiente en arrestine 1 [4] ou en GRK1 [5] ont montré qu’une exposition trop longue à la lumière conduit à la dégénérescence de la rétine.
Fuchs et ses collaborateurs [6] ont été les premiers à identifier une délétion homozygote du gène de l’arrestine 1 chez des patients, japonais, atteints de la maladie d’Oguchi : cette délétion, qui porte sur une adénine de l’exon 11, conduit à un décalage du cadre de lecture et à un arrêt prématuré de la traduction. Par ailleurs, des mutations du gène de la GRK1 ont été décrites chez trois patients qui, en revanche, avaient un gène de l’arrestine 1 intact [7] : deux cas présentaient une délétion homozygote de l’exon 5, tandis que le 3e cas était hétérozygote, avec une mutation de type faux sens, dans l’exon 5, aboutissant à la substitution de la valine 380 par un acide aspartique, et une délétion de 4 paires de bases dans l’exon 7, créant une protéine tronquée dans sa partie carboxyterminale. Les protéines GRK1 exprimant ces mutations sont totalement dénuées d’activité enzymatique.
Ces résultats démontrent clairement qu’une stimulation excessive de la rhodopsine, provoquée par une abolition de la fonction de la GRK1 ou de l’arrestine 1, entraîne une situation pathologique touchant le système visuel. Un rétablissement des fonctions de la GRK1 ou de l’arrestine 1 provoquerait, sans nul doute, un retour à l’état normal (Tableau I).
Tableau I
Implication des GRK et des arrestines en pathologie et piste thérapeutique.

Figure 2
Protéines de liaison et rôles des β-arrestines.

Trois grands rôles sont attribués aux β-arrestines : la désensibilisation, l’internalisation des récepteurs couplés aux protéines G (RCPG) et le déclenchement de nouvelles voies de signalisation. Les mécanismes membranaires de la désensibilisation (Figure 1) font intervenir la liaison des β-arrestines aux RCPG phosphorylés et aux isoenzymes de la phosphodiestérase 4 (PDE4). L’internalisation permet la dégradation lysosomiale du récepteur, ou son recyclage après déphosphorylation par une protéine phosphatase. Les protéines liant les β-arrestines et participant à l’endocytose sont la clathrine, les protéines AP2 (adaptor protein 2), ARF6 (ADP ribozylation factor 6), ARNO (ARF nucleotide exchange factor), Mdm2 (ubiquitine ligase E3), NSF (N-ethylmaleimide-sensitive fusion protein), Dvl2 (Dishevelled 2) et les PI (phospho-inositides). Les protéines liant les β-arrestines et participant au déclenchement de nouvelles voies de signalisation sont les protéines de la famille Src (c-Src, Yes, Fgr, Hck), certaines MAPK (mitogen-activated protein kinases, ERK2, c-Raf1, JNK3 et ASK1), la protéine kinase B (Akt), un facteur d’échange des nucléotides à guanine (Ral-GDS), IκBα (inhibiteur du facteur de transcription NF-κB), la PP2A (protéine phosphatase 2A) et Dvl1/2 (Dishevelled 1/2). L : ligand.
Pathologies accompagnant un défaut de régulation des récepteurs β-adrénergiques
Les récepteurs β-adrénergiques possèdent une structure à sept domaines transmembranaires couplés à l’adénylyl cyclase par l’intermédiaire d’une protéine Gs, entraînant la synthèse d’AMPc. Les études réalisées chez des souris transgéniques ont montré le rôle prépondérant de la GRK2, de la GRK5 et de l’arrestine 2 dans la régulation des récepteurs β-adrénergiques cardiaques. Chez l’homme, une augmentation de la GRK2 est observée dans la défaillance cardiaque chronique (DCC) [8], l’hypertension [9] et la mucoviscidose [10], ainsi que dans d’autres pathologies cardiaques conduisant à la dilatation du ventricule gauche. Dans tous les cas, l’augmentation de l’activité GRK est corrélée à une baisse de la synthèse de l’AMPc ou à la diminution de la fonction du récepteur stimulé par un agoniste β dans les tissus concernés.
Afin d’expliquer la baisse de la signalisation, Gros et ses collaborateurs [9] ont exploré l’expression des protéines impliquées dans la désensibilisation des récepteurs β-adrénergiques des lymphocytes de personnes jeunes (20-36 ans) atteintes d’hypertension (l’expression de la GRK2 suit un parallèle entre les cellules musculaires lisses des vaisseaux et les lymphocytes) : les résultats ont montré une augmentation de l’expression de la GRK2, accompagnée d’une hausse de l’activité enzymatique des GRK chez les sujets hypertendus en comparaison des sujets normaux. Cette variation de la GRK2 est spécifique, puisqu’aucune modification de la GRK5, de la GRK6, de la PKA ou des β-arrestines 1 et -2 n’a été détectée. De plus, une corrélation positive est observée entre l’expression de la GRK2 et la valeur de la pression artérielle systolique.
La mise au point de souris transgéniques surexprimant, de façon ciblée, la GRK2 au niveau vasculaire a permis d’examiner le rôle de cette enzyme sur la régulation de la pression artérielle in vivo [11]. La stimulation des récepteurs β-adrénergiques produit un effet vasodilatateur atténué chez les souris transgéniques par rapport aux souris contrôles. De plus, la surexpression vasculaire de la GRK2 provoque une augmentation spontanée de la pression artérielle, avec un épaississement de la paroi aortique.
En conclusion, la GRK2 est un élément important dans la réponse des récepteurs β-adrénergiques vasculaires et dans la régulation de la pression artérielle (Tableau I). Cependant, le rôle de cette enzyme est probablement multiple, puisque la phosphorylation des canaux sodiques épithéliaux par la GRK2 a été avancée pour expliquer certaines causes d’hypertension d’origine rénale [12].
Baisse de l’expression des GRK dans l’arthrite rhumatoïde
Une forte diminution de la GRK2 (d’environ 55 %) et de la GRK6 (d’environ 60 %) est observée dans les cellules mononucléées sanguines de patients atteints d’arthrite rhumatoïde, par rapport aux cellules de sujets témoins [13]. Cette diminution permet d’expliquer l’augmentation de la synthèse de l’AMPc en réponse à un agoniste des récepteurs β2-adrénergiques dans les lymphocytes. De nombreux récepteurs couplés aux protéines G (récepteurs des prostaglandines, de la substance P ou des chimiokines, récepteurs β2-adrénergiques) participent au processus inflammatoire de l’arthrite. De plus, les cytokines dont la synthèse est augmentée dans l’arthrite rhumatoïde (interféron γ et interleukine 6) inhibent la production de GKR2 dans des cellules en culture, ce qui pourrait expliquer les effets observés in vivo. Aucune variation de la GRK5 ni des arrestines 2 et -3 n’a été notée dans les lymphocytes de patients atteints d’arthrite rhumatoïde [13]. Ces résultats ont été confirmés chez le rat présentant une arthrite induite par l’injection de substances inflammatoires [14]. Chez l’animal, les niveaux d’expression de la GRK2, de la GRK3 et de la GRK6 varient en fonction de l’intensité des signes cliniques de l’arthrite adjuvante, avec une baisse lors du pic inflammatoire et une remontée vers la normale lors de la phase de rémission de la maladie. La diminution de la GRK2 semble spécifique des cellules impliquées dans la réponse inflammatoire (cellules T CD4+ et cellules B), puisqu’aucun changement n’a été observé dans le coeur ou l’hypophyse.
Ces résultats chez l’animal et chez l’homme suggèrent que la baisse des GRK serait une conséquence du processus inflammatoire, car la diminution de l’expression de ces kinases suit les signes cliniques de la maladie, au lieu de les précéder. Par ailleurs, des expériences menées chez des souris GRK2+/- et GRK6-/- ont montré le rôle de ces enzymes dans le chimiotactisme in vitro des lymphocytes T [15] et des polynucléaires neutrophiles [16], suggérant que la baisse de ces GRK pourrait être impliquée dans la migration des cellules immunitaires lors de l’arthrite rhumatoïde.
Régulation des récepteurs dopaminergiques dans l’hypertension et la maladie de Parkinson
Cinq récepteurs distincts couplés aux protéines G sont activés par la dopamine. Sur la base d’une similitude de séquences et de fonctions, ces récepteurs ont été subdivisés en deux sous-groupes : les récepteurs D1-like (D1 et D5) et les récepteurs D2-like (D2, D3 et D4).
De nombreux arguments ont permis d’associer l’hypertension et la désensibilisation des récepteurs D1 dopaminergiques par la GRK4 : la signalisation par les récepteurs D1 dopaminergiques est fortement diminuée dans l’hypertension essentielle, conduisant à la baisse de l’effet natriurétique de la dopamine au niveau du tube contourné proximal du rein ; la GRK4 désensibilise in vitro les récepteurs D1, et les différentes isoformes de la GRK4 (α, β, γ, δ) sont exprimées au niveau du tube contourné proximal [17] ; une association est observée entre la présence du variant allélique A486V de la GRK4γ et l’hypertension essentielle chez l’homme [18] ; enfin, ces résultats ont été confirmés chez l’animal, par la mise au point d’animaux spontanément hypertendus après transfection du variant A142V de la GRK4γ [17]. Les variants A486V et A142V stimulent l’activité enzymatique de la GRK4γ et une inhibition de celle-ci pourrait représenter une autre voie thérapeutique de l’hypertension (Tableau I).
La maladie de Parkinson est liée à une dégénérescence des neurones dopaminergiques de la voie nigrostriatale du cerveau. La redistribution des récepteurs dopaminergiques (D1, D2 et D3) qui suit cette dégénérescence serait à l’origine des troubles moteurs et de la dyskinésie caractéristiques de la maladie de Parkinson. Une étude réalisée chez le macaque a montré une augmentation de l’arrestine 2 et de la GRK6 dans le cerveau de singes porteurs d’une maladie de Parkinson induite par un agent neurotoxique, le MPTP[1] [19]. L’augmentation de la GRK6 contribuerait au blocage des récepteurs dopaminergiques centraux, ce que corrobore la stimulation des effets locomoteurs des psychostimulants (cocaïne et amphétamine), attribuée à l’activation indirecte des récepteurs D2-like chez des souris transgéniques dépourvues de GRK6. Ainsi, l’inhibition de l’activité GRK6 pourrait être envisagée pour réguler les mouvements des patients atteints de la maladie de Parkinson (Tableau I).
Désensibilisation des RCPG et maladies psychiatriques
Une augmentation de la GRK2 membranaire est observée dans la région du cortex préfrontal de personnes dépressives et décédées par suicide [20]. Inversement, l’expression de la GRK2 est réduite après un traitement antidépresseur [21]. De même, une variation du taux de GRK2 dans les plaquettes sanguines de personnes atteintes d’une dépression majeure est contrebalancée par un traitement antidépresseur utilisant un antagoniste des récepteurs α2-adrénergiques. Par ailleurs, une augmentation de l’arrestine 2 est observée dans le cortex et l’hippocampe de rats sous traitement antidépresseur. Chez l’homme, l’expression de l’arrestine 2 dans les cellules mononuclées du sang est inversement corrélée à la sévérité de la dépression [22], la normalisation de son taux précédant l’amélioration des signes cliniques de la maladie [23]. L’ensemble de ces résultats suggère un lien probable entre les récepteurs α2A-adrénergiques et la GRK2 chez les sujets atteints de dépression majeure, et souligne le rôle de la GRK2 et de l’arrestine 2 comme marqueurs biochimiques potentiels dans la réponse au traitement.
GRK/arrestine et addiction aux opiacés
Trois types de récepteurs des opiacés (μ, κ et δ) appartiennent à la famille des RCPG ; parmi eux, le récepteur de type μ joue un rôle prépondérant dans l’action de la morphine, d’où l’intérêt de l’étude de sa désensibilisation dans les phénomènes de tolérance et de dépendance aux substances opiacées. Des variations d’expression des GRK 2, -3, -5 et -6, ainsi que des arrestine 2 et -3, ont été observées dans le cerveau d’hommes ou de rats ayant reçu des opiacés ou leurs antagonistes sous forme d’une administration aiguë ou chronique [24]. Ces variations s’accordent avec la démonstration du rôle physiologique de l’arrestine 3 dans l’effet analgésique et la tolérance à la morphine chez des souris dépourvues de cette protéine [25]. Chez la souris, toujours, la GRK3 joue un rôle important dans la tolérance impliquant les récepteurs de type μ [26] et κ [27]. Les phénomènes de dépendance ne semblent pas affectés chez la souris invalidée pour le gène de l’arrestine 3, mais la baisse de la tolérance suggère une diminution de l’overdose(Tableau I).
GRK/arrestine et pathologies hormonodépendantes
Une étude portant sur le cancer de l’ovaire a montré des variations de la GRK2 et des isoformes de la GRK4 aussi bien dans une lignée cancéreuse que dans des échantillons tissulaires de tumeurs provenant de cellules de la granulosa. Les auteurs de cette étude concluaient au rôle possible des GRK dans la pathogénie de ce cancer, impliquant la régulation de l’effet prolifératif de la follitropine [28].
Une augmentation de l’activité GRK2 et une baisse de l’expression de la GRK5 ont été observées dans les cancers différenciés de la thyroïde [29] ; les variations de la GRK5 sont corrélées à la diminution de la désensibilisation des récepteurs de la thyrotropine (TSH), ou encore à l’augmentation de la synthèse de l’AMPc dans ces tissus, sur lesquels la TSH a un effet mitogène.
Plusieurs travaux ont montré le rôle antiprolifératif d’une surexpression de la GRK2 [30] ou de son domaine carboxyterminal (blocage du complexe βγ des protéines G) [31] sur la croissance de certaines lignées en culture (Tableau I). De plus, la prolifération d’un cancer de la prostate chez la souris a été fortement ralentie par la transfection du domaine carboxyterminal de la GRK2 [31].
Des variations de l’expression des GRK et des β-arrestines [32] ont été observées dans des nodules bénins de la thyroïde, suggérant un dysfonctionnement de la régulation du signal TSH dans ces tissus.
Modification thérapeutique de la GRK2 ou de l’arrestine 3 chez l’animal
GRK2 dans la défaillance cardiaque chronique, la sclérose en plaque et le diabète de type 2
Chez l’homme, la défaillance cardiaque chronique s’accompagne d’une baisse du nombre de récepteurs β-adrénergiques et d’une augmentation de la GRK2 au niveau du coeur. Plusieurs modèles d’animaux ont permis de confirmer le rôle important de la GRK2 dans la régulation des récepteurs β-adrénergiques, et d’identifier la GRK2 comme une cible potentielle dans le traitement de cette maladie. Ainsi, des souris porteuses d’une inactivation du gène codant pour la protéine musculaire LIM présentent une hypertrophie cardiaque, avec un défaut de transduction des récepteurs β-adrénergiques et une augmentation de la GRK2 très similaire à ce qui est observé chez l’homme au cours de la défaillance cardiaque. Le croisement de ces souris avec une lignée exprimant un peptide inhibiteur de la GRK2 (peptide carboxyterminal de la GRK2) a donné naissance à des animaux présentant une baisse de l’activité GRK2 et une amélioration de la fonction contractile du coeur [33]. Chez le lapin, l’utilisation d’un vecteur viral pour délivrer in vivo le peptide inhibiteur de la GRK2 a démontré son efficacité, en retardant les signes cliniques de la défaillance cardiaque après un infarctus du myocarde [34].
Une baisse de la GRK2 a été observée dans les cellules mononuclées sanguines de patients atteints de sclérose en plaques, avec une diminution encore plus prononcée durant la phase de rémission de cette maladie inflammatoire chronique touchant la gaine de myéline [35]. L’absence de rechute ainsi qu’une évolution plus favorable des signes cliniques dans un modèle animal de sclérose en plaques dont la GRK2 a été génétiquement diminuée de 50 % [35] suggère que l’inhibition de la GRK2 pourrait devenir une alternative au traitement de cette maladie (Tableau I).
Le troisième exemple concerne l’effet antidiabétique de l’inhibition in vivo de l’activité GRK2/3 par de petits peptides dérivés du site actif des kinases [36] : dans un modèle animal (gerbilles Psammomys obesus) de diabète de type 2, l’injection intrapéritonéale des peptides inhibiteurs de la GRK2 et de la GRK3 produit une baisse de la glycémie de 72 % de sa valeur avant traitement. Bien que le rôle exact des GRK dans la régulation de la glycémie demeure inconnu, cette étude démontre que la modulation de l’activité GRK2/3 peut être efficace dans le traitement du diabète de type 2, et que les peptides porteurs d’une chaîne d’acide gras sont peu toxiques chez l’animal.
Arrestine 3 et développement de l’asthme allergique
La souris sensibilisée à l’ovalbumine a été utilisée comme modèle animal de l’asthme allergique. Chez ces souris, dépourvues d’arrestine 3 par manipulation génétique, la migration des lymphocytes T (T helper 2) ainsi que les manifestations inflammatoires de l’asthme sont très réduites [37]. Les mécanismes physiologiques permettant d’expliquer le rôle de l’arrestine 3 dans la réponse immune à un allergène, non encore élucidés, pourraient faire intervenir la régulation des RCPG participant au chimiotactisme : les lymphocytes spléniques de souris dépourvues d’arrestine 3 présentent en effet un index chimiotactique fortement diminué, in vitro, vis-à-vis du facteur de migration SDF (stromal cell-derived factor-1α), ligand du récepteur CXCR4 (chemokine CXC motif receptor 4) [38]. Ces résultats ont également été obtenus sur des cellules non immunes surexprimant les récepteurs CXCR4 et CCR5 (chemokine CC motif receptor 5) [39] : dans ces cellules, une baisse de 80 % de l’expression de l’arrestine 3, par la technique d’interférence par l’ARN, diminue le pouvoir de migration cellulaire, une piste pour contrôler les manifestations inflammatoires de l’asthme allergique (Tableau I).
Conclusions et perspectives
L’importance physiologique du couple GRK/arrestine est mise en évidence dans de nombreuses situations pathologiques où la réponse des RCPG est altérée. À côté des maladies liées à des mutations génétiques comme la maladie d’Oguchi et la prédisposition à l’hypertension, un certain nombre d’états pathologiques a été associé à des variations d’expression des GRK et des arrestines. La mise au point de modèles de souris présentant des problèmes visuels ou spontanément hypertendues confirme bien le rôle causal, sinon majeur, des GRK1, -2 et -4γ, ainsi que de l’arrestine 1, dans ces pathologies.
De plus, l’amélioration des signes cliniques de la défaillance cardiaque chronique, de la sclérose en plaque, du diabète ou de l’asthme allergique par la modification de l’activité ou de l’expression de la GRK2 ou de l’arrestine 3 démontre l’importance de ces protéines dans la physiopathologie de ces maladies, et ouvre les portes à une éventuelle piste thérapeutique. Bien que l’utilisation de petits peptides inhibiteurs de l’activité GRK2/3 soit une voie prometteuse, l’arsenal thérapeutique pour envisager des essais chez l’homme reste pauvre, à moins que la thérapie génique fasse des progrès substantiels. La question de la place de ces thérapeutiques potentielles parmi celles déjà existantes est importante, notamment dans l’insuffisance cardiaque où les β-bloquants ont fait la preuve de leur efficacité. En fait, ces deux thérapies pourraient avoir des effets complémentaires, puisqu’elles agissent sur la même voie de signalisation mais à des niveaux différents : les β-bloquants antagonisent l’effet des catécholamines sur le récepteur, tandis que l’inhibition de la GRK2 préserve le couplage normal du récepteur avec les protéines G, permettant ainsi une adaptation plus facile à l’exercice ou à une période de stress.
Parties annexes
Remerciements
Nous remercions la Ligue contre le cancer, comité de la Vienne, pour l’aide financière qu’elle apporte à notre laboratoire.
Note
-
[1]
1-méthyl-4-phényl-1,2,3,6-tétrahydropyridine
Références
- 1. Krupnick JG, Benovic JL. The role of receptor kinases and arrestins in G protein-coupled receptor regulation. Annu Rev Pharmacol Toxicol 1998 ; 38 : 289-319.
- 2. Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta-arrestins. Science 2005 ; 308 : 512-7.
- 3. Metaye T, Gibelin H, Perdrisot R, Kraimps JL. Pathophysiological roles of G-protein-coupled receptor kinases. Cell Signal 2005 ; 17 : 917-28.
- 4. Chen J, Simon MI, Matthes MT, et al. Increased susceptibility to light damage in an arrestin knockout mouse model of Oguchi disease (stationary night blindness). Invest Ophthalmol Vis Sci 1999 ; 40 : 2978-82.
- 5. Chen CK, Burns ME, Spencer M, et al. Abnormal photoresponses and light-induced apoptosis in rods lacking rhodopsin kinase. Proc Natl Acad Sci USA 1999 ; 96 : 3718-22.
- 6. Fuchs S, Nakazawa M, Maw M, et al. A homozygous 1-base pair deletion in the arrestin gene is a frequent cause of Oguchi disease in Japanese. Nat Genet 1995 ; 10 : 360-2.
- 7. Yamamoto S, Sippel KC, Berson EL, Dryja TP. Defects in the rhodopsin kinase gene in the Oguchi form of stationary night blindness. Nat Genet 1997 ; 15 : 175-8.
- 8. Ungerer M, Bohm M, Elce JS, et al. Altered expression of beta-adrenergic receptor kinase and beta 1-adrenergic receptors in the failing human heart. Circulation 1993 ; 87 : 454-63.
- 9. Gros R, Benovic JL, Tan CM, Feldman RD. G-protein-coupled receptor kinase activity is increased in hypertension. J Clin Invest 1997 ; 99 : 2087-93.
- 10. Mak JC, Chuang TT, Harris CA, Barnes PJ. Increased expression of G protein-coupled receptor kinases in cystic fibrosis lung. Eur J Pharmacol 2002 ; 436 : 165-72.
- 11. Eckhart AD, Ozaki T, Tevaearai H, et al. Vascular-targeted overexpression of G protein-coupled receptor kinase-2 in transgenic mice attenuates beta-adrenergic receptor signaling and increases resting blood pressure. Mol Pharmacol 2002 ; 61 : 749-58.
- 12. Dinudon A, Fotia AB, Lefkowitz RJ, et al. The kinase GRK2 regulates Nedd4/Nedd4-2-dependent control of epithelial Na+ channels. Proc Natl Acad Sci USA 2004 ; 101 : 11886-90.
- 13. Lombardi MS, Kavelaars A, Schedlowski M, et al. Decreased expression and activity of G-protein-coupled receptor kinases in peripheral blood mononuclear cells of patients with rheumatoid arthritis. FASEB J 1999 ; 13 : 715-25.
- 14. Lombardi MS, Kavelaars A, Cobelens PM, et al. Adjuvant arthritis induces down-regulation of G protein-coupled receptor kinases in the immune system. J Immunol 2001 ; 166 : 1635-40.
- 15. Vroon A, Heijnen CJ, Lombardi MS, et al. Reduced GRK2 level in T cells potentiates chemotaxis and signaling in response to CCL4. J Leukoc Biol 204 ; 75 : 901-9.
- 16. Vroon A, Heijnen CJ, Raatgever R, et al. GRK6 deficiency is associated with enhanced CXCR4-mediated neutrophil chemotaxis in vitro and impaired responsiveness to G-CSF in vivo. J Leukoc Biol 2004 ; 75 : 698-704.
- 17. Felder RA, Sanada H, Xu J, et al. G protein-coupled receptor kinase 4 gene variants in human essential hypertension. Proc Natl Acad Sci USA 2002 ; 99 : 3872-7.
- 18. Speirs HJ, Katyk K, Kumar NN, et al. Association of G-protein-coupled receptor kinase 4 haplotypes, but not HSD3B1 or PTP1B polymorphisms, with essential hypertension. J Hypertens 2004 ; 22 : 931-6.
- 19. Bezard E, Gross CE, Qin L, et al. L-DOPA reverses the MPTP-induced elevation of the arrestin2 and GRK6 expression and enhanced ERK activation in monkey brain. Neurobiol Dis 2005 ; 18 : 323-35.
- 20. Garcia-Sevilla JA, Escriba PV, Ozaita A, et al. Up-regulation of immunolabeled alpha2A-adrenoceptors, Gi coupling proteins, and regulatory receptor kinases in the prefrontal cortex of depressed suicides. J Neurochem 1999 ; 72 : 282-91.
- 21. Grange-Midroit M, Garcia-Sevilla JA, Ferrer-Alcon M, et al. Regulation of GRK 2 and 6, beta-arrestin-2 and associated proteins in the prefrontal cortex of drug-free and antidepressant drug-treated subjects with major depression. Brain Res Mol Brain Res 2003 ; 111 : 31-41.
- 22. Avissar S, Matuzany-Ruban A, Tzukert K, Schreiber G. Beta-arrestin-1 levels : reduced in leukocytes of patients with depression and elevated by antidepressants in rat brain. Am J Psychiatry 2004 ; 161 : 2066-72.
- 23. Matuzany-Ruban A, Avissar S, Schreiber G. Dynamics of beta-arrestin1 protein and mRNA levels elevation by antidepressants in mononuclear leukocytes of patients with depression. J Affect Disord 2005 ; 88 : 307-12.
- 24. Ferrer-Alcon M, La Harpe R, Garcia-Sevilla JA. Decreased immunodensities of micro-opioid receptors, receptor kinases GRK 2/6 and beta-arrestin-2 in postmortem brains of opiate addicts. Brain Res Mol Brain Res 2004 ; 121 : 114-22.
- 25. Bohn LM, Gainetdinov RR, Lin FT, et al. Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature 2000 ; 408 : 720-3.
- 26. Terman GW, Jin W, Cheong YP, et al. G-protein receptor kinase 3 (GRK3) influences opioid analgesic tolerance but not opioid withdrawal. Br J Pharmacol 2004 ; 141 : 55-64.
- 27. Xu M, Petraschka M, McLaughlin JP, et al. Neuropathic pain activates the endogenous kappa opioid system in mouse spinal cord and induces opioid receptor tolerance. J Neurosci 2004 ; 24 : 4576-84.
- 28. King DW, Steinmetz R, Wagoner HA, et al. Differential expression of GRK isoforms in nonmalignant and malignant human granulosa cells. Endocrine 2003 ; 22 : 135-42.
- 29. Metaye T, Menet E, Guilhot J, Kraimps JL. Expression and activity of G protein-coupled receptor kinases in differentiated thyroid carcinoma. J Clin Endocrinol Metab 2002 ; 87 : 3279-86.
- 30. Iacovelli L, Capobianco L, D’Ancona GM, et al. Regulation of lysophosphatidic acid receptor-stimulated response by G-protein-coupled receptor kinase-2 and beta-arrestin1 in FRTL-5 rat thyroid cells. J Endocrinol 2002 ; 174 : 103-10.
- 31. Bookout AL, Finney AE, Guo R, et al. Targeting Gbetagamma signaling to inhibit prostate tumor formation and growth. J Biol Chem 2003 ; 278 : 37569-73.
- 32. Voigt C, Holzapfel H-P, Paschke R. Expression of β-arrestins in toxic and cold thyroid nodules. FEBS Lett 2000 ; 486 : 208-12.
- 33. Rockman HA, Chien KR, Choi DJ, et al. Expression of a beta-adrenergic receptor kinase 1 inhibitor prevents the development of myocardial failure in gene-targeted mice. Proc Natl Acad Sci USA 1998 ; 95 : 7000-5.
- 34. White DC, Hata JA, Shah AS, et al. Preservation of myocardial β-edrenergic receptor signaling delays the development of heart failure after myocardial infarction. Proc Natl Acad Sci USA 2000 ; 97 : 5428-33.
- 35. Vroon A, Kavelaars A, Limmroth V, et al. G protein-coupled receptor kinase 2 in multiple sclerosis and experimental autoimmune encephalomyelitis. J Immunol 2005 ; 174 : 4400-6.
- 36. Anis Y, Leshem O, Reuveni H, et al. Antidiabetic effect of novel modulating peptides of G-protein-coupled kinase in experimental models of diabetes. Diabetologia 2004 ; 47 : 1232-44.
- 37. Walker JK, Fong AM, Lawson BL, et al. Beta-arrestin-2 regulates the development of allergic asthma. J Clin Invest 2003 ; 112 : 566-74.
- 38. Fong AM, Premont RT, Richardson RM, et al. Defective lymphocyte chemotaxis in beta-arrestin2- and GRK6-deficient mice. Proc Natl Acad Sci USA 2002 ; 99 : 7478-83.
- 39. Sun Y, Cheng Z, Ma L, Pei G. Beta-arrestin2 is critically involved in CXCR4-mediated chemotaxis, and this is mediated by its enhancement of p38 MAPK activation. J Biol Chem 2002 ; 277 : 49212-9.
Liste des figures
Figure 1
Mécanismes membranaires de la désensibilisation du récepteur β2-adrénergique.
Ce récepteur, qui a fait l’objet d’études approfondies, est considéré comme un prototype dans la régulation de la transduction du signal couplée à l’activation de l’adénylyl cyclase. La fixation du ligand sur le récepteur β2-adrénergique provoque la synthèse de l’AMPc par l’adénylyl cyclase activée par la sous unité Gαs liée au GTP. Dans les instants qui suivent, la GRK2 est recrutée à la membrane plasmique, où elle fixe la sous-unité βγ et les phospholipides. La localisation de la GRK2 lui permet de phosphoryler des résidus sérine et thréonine de la partie carboxyterminale du récepteur (étape 1). Le récepteur β2-adrénergique phosphorylé par la GRK2 a une affinité accrue pour la β-arrestine 2, qui accentue le processus de désensibilisation en empêchant le couplage d’une nouvelle protéine Gs. De plus, la liaison de la phosphodiestérase 4 (PDE4) à la β-arrestine 2 contribue à la transformation de l’AMPc en AMP, et limite ainsi une stimulation excessive de la cellule par le second messager (étape 2).
Figure 2
Protéines de liaison et rôles des β-arrestines.
Trois grands rôles sont attribués aux β-arrestines : la désensibilisation, l’internalisation des récepteurs couplés aux protéines G (RCPG) et le déclenchement de nouvelles voies de signalisation. Les mécanismes membranaires de la désensibilisation (Figure 1) font intervenir la liaison des β-arrestines aux RCPG phosphorylés et aux isoenzymes de la phosphodiestérase 4 (PDE4). L’internalisation permet la dégradation lysosomiale du récepteur, ou son recyclage après déphosphorylation par une protéine phosphatase. Les protéines liant les β-arrestines et participant à l’endocytose sont la clathrine, les protéines AP2 (adaptor protein 2), ARF6 (ADP ribozylation factor 6), ARNO (ARF nucleotide exchange factor), Mdm2 (ubiquitine ligase E3), NSF (N-ethylmaleimide-sensitive fusion protein), Dvl2 (Dishevelled 2) et les PI (phospho-inositides). Les protéines liant les β-arrestines et participant au déclenchement de nouvelles voies de signalisation sont les protéines de la famille Src (c-Src, Yes, Fgr, Hck), certaines MAPK (mitogen-activated protein kinases, ERK2, c-Raf1, JNK3 et ASK1), la protéine kinase B (Akt), un facteur d’échange des nucléotides à guanine (Ral-GDS), IκBα (inhibiteur du facteur de transcription NF-κB), la PP2A (protéine phosphatase 2A) et Dvl1/2 (Dishevelled 1/2). L : ligand.
Liste des tableaux
Tableau I
Implication des GRK et des arrestines en pathologie et piste thérapeutique.