Résumés
Résumé
L’endothéline-1 (ET-1) et l’angiotensine II (AngII) sont des peptides vasoactifs, mais aussi mitogènes et pro-angiogéniques. Tous deux exercent leurs actions par l’intermédiaire de récepteurs couplés aux protéines G : ETA-R et ETB-R pour ET-1 ; AT1R et AT2R pour AngII. L’expression des composants des systèmes ET-1 et AngII dans diverses tumeurs présente généralement une ou plusieurs des caractéristiques suivantes : surexpression du peptide et/ou du récepteur, modification du sous-type de récepteur exprimé et localisation nucléaire du récepteur. ET-1 et AngII agissent sur les différentes étapes de la progression tumorale, et l’utilisation d’antagonistes spécifiques de leurs récepteurs, ou d’inhibiteurs de leur synthèse, est efficace pour ralentir la croissance tumorale in vitro et in vivo dans différents modèles animaux. Des essais cliniques utilisant des antagonistes d’ETA-R donnent des résultats encourageants pour le traitement antitumoral, et une approche similaire ayant pour objectif de bloquer ETB-R ou AT1R est envisageable. De plus, une thérapie combinée ciblant les deux systèmes, ET-1 et AngII, pourrait se révéler bénéfique pour le traitement de tumeurs fortement angiogéniques.
Summary
Endothelin-1 (ET-1) and angiotensin II (AngII), two potent vasoactive peptides involved in the regulation of cardiovascular homeostasis, also induce mitogenic and pro-angiogenic responses in vitro and in vivo. Both peptides are produced by cleavage of inactive precursors by metalloproteases (endothelin-converting enzyme and angiotensin-converting enzyme, respectively) and activate two subtypes of membrane receptors (ETA-R and ETB-R for ET-1, AT1R and AT2R for AngII) that all belong to the superfamily of G-protein coupled receptors. There is increasing evidence that ETA-R, ETB-R and AT1R are expressed in a variety of cancer cells and tissues, and may play a role on tumor growth, angiogenesis and invasion in vivo. This review summarizes the similarities and differences between the ET-1 and AngII systems with regard to their reported effects on various aspects of cancer. In addition to being expressed on vascular endothelium, ET-1 and AngII receptors participate in tumor angiogenesis through the production of the angiogenic factor VEGF. Furthermore, recent clinical studies indicate that a selective ETA-R antagonist has beneficial effects in prostate cancer, suggesting that a similar approach using ETB-R and AT1R blockers might be envisioned. Experimental data presented here suggest that a combined therapy targeting both ET-1 and AngII systems may prove valuable for future treatments of highly angiogenic tumors.
Corps de l’article
Expression des systèmes endothéline-1 et angiotensine II dans les tumeurs
Des similitudes dans les deux systèmes
L’endothéline-1 (ET-1), l’isopeptide le plus étudié du système endothéline, et l’angiotensine II (AngII), le peptide biologiquement actif du système rénine-angiotensine, ont d’abord été caractérisés pour leur vasoactivité. Depuis, de nombreuses études ont illustré leurs effets sur diverses fonctions cellulaires, dont la prolifération. L’ET-1 (21 acides aminés) est essentiellement sécrétée par les cellules endothéliales, alors que l’AngII (8 acides aminés) est produite dans la circulation (endocrine), mais aussi localement (paracrine), notamment à la surface des cellules endothéliales. Ces deux peptides sont produits par un mécanisme similaire, faisant intervenir la coupure enzymatique de précurseurs inactifs par une métalloprotéase (Figure 1) : l’enzyme de conversion d’ET-1 (ECE-1) clive la « Big ET-1 », tandis que l’enzyme de conversion de l’AngI (ECA) clive l’AngI.
Les deux peptides ET-1 et AngII ont de nombreuses cibles cellulaires, parmi lesquelles les cellules endothéliales et les cellules musculaires lisses des vaisseaux. Ils exercent tous deux leurs actions par l’intermédiaire de récepteurs appartenant à la superfamille des récepteurs à sept domaines transmembranaires couplés aux protéines G hétérotrimériques (RCPG). Dans le système ET-1, deux sous-types de récepteurs ont été identifiés, ETA-R et ETB-R, qui transmettent des effets vasoactifs opposés : vasoconstriction couplée à ETA-R dans les cellules musculaires lisses, vasodilation couplée à ETB-R dans les cellules endothéliales. En revanche, tous deux sont capables de relayer l’effet mitogène d’ET-1 [1, 2]. AngII se lie également à deux récepteurs : AT1R, exprimé de façon prédominante chez l’adulte, et AT2R, exprimé chez le foetus et dans des situations de remodelage tissulaire. Les effets physiologiques d’AngII sont majoritairement attribués à AT1R, AT2R étant considéré comme un récepteur régulateur. Comme ET-1, AngII régule positivement la prolifération et la migration cellulaires par l’intermédiaire d’AT1R, tandis qu’AT2R exerce un contrôle négatif sur ces fonctions [3].
Expression des systèmes ET-1 et AngII dans les tumeurs
Pour la première fois en 1995, une étude a suggéré qu’ET-1 pouvait être impliqué dans le cancer de la prostate [4]. Depuis, une expression de plusieurs des composants du système ET-1, et plus récemment du système AngII, a été mise en évidence dans différentes cellules cancéreuses (Tableau I) [5-10]. Outre une élévation de la synthèse du peptide, une régulation positive de l’expression du récepteur est couramment observée. C’est ainsi qu’une augmentation de l’expression de composants du système ET-1 (prépro-ET-1, ECE-1, ETA-R) a pu être reliée à l’invasivité de certaines tumeurs (adénome colorectal, cancers du sein et de la prostate). Il en est de même pour l’expression du système AngII, notamment dans le cas de cancers de la peau ou de carcinomes cervicaux.
Tableau I
Expression des récepteurs de l’endothéline 1 et de l’angiotensine II dans différents tissus tumoraux et lignées cellulaires tumorales.

+ : exprimé ; ++ : surexprimé ; - : non exprimé ; +/- : faiblement détecté. CDIS : carcinome ductal in situ.
Figure 1
Similitudes entre les étapes de synthèse de l’endothéline 1 (ET-1) et de l’angiotensine II (AngII) et leurs modes d’action.
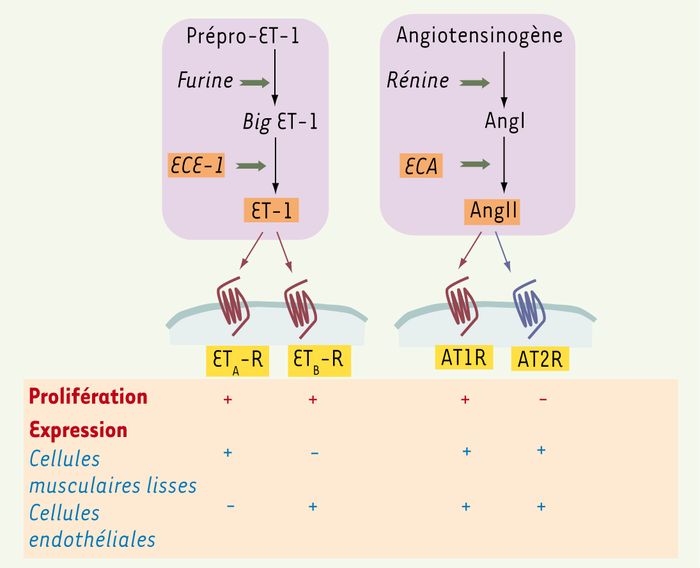
ET-1 et AngII sont tous deux des produits de clivage de précurseurs inactifs par une métalloprotéase membranaire, respectivement l’ECE-1 (enzyme de conversion d’ET-1) et l’ECA (enzyme de conversion de l’angiotensine I). Ils agissent par l’intermédiaire de récepteurs appartenant à la même superfamille des récepteurs couplés aux protéines G (RCPG) : ETA-R et ETB-R pour ET-1, AT1R et AT2R pour AngII. Alors qu’ETA-R, ETB-R et AT1R sont couplés positivement à la prolifération, AT2R a une action antiproliférative. L’endothélium vasculaire est une des cibles communes d’ET-1 et AngII.
Les récepteurs d’AngII surexprimés dans les cellules tumorales sont essentiellement du sous-type AT1R, même si une augmentation de l’expression d’AT2R a été observée dans les cancers du sein, suggérant une éventuelle participation des deux sous-types de récepteurs à la progression tumorale. Pour le système ET-1, la surexpression des récepteurs ETA-R et ETB-R est observée de manière différentielle selon le type cellulaire (Tableau I), la surexpression d’ETA-R étant plus particulièrement notée dans les tumeurs d’origine épithéliale (carcinomes). De manière intéressante, les cellules tumorales expriment le plus souvent un sous-type de récepteur d’ET-1 différent de celui qui est exprimé par le tissu normal correspondant : ainsi, alors que les astrocytes et les mélanocytes expriment respectivement les récepteurs ETB-R et ETA-R, les glioblastomes et les mélanomes expriment, à l’inverse, ETA-R et ETB-R, un profil d’expression correspondant aux cellules non différenciées [10-12].
Récemment, nous avons observé une expression nucléaire des récepteurs d’ET-1 dans les cellules tumorales de gliomes : ETB-R dans les oligodendrogliomes, et ETA-R dans les glioblastomes [10]. De même, une localisation nucléaire de l’AT1R a été mise en évidence dans des hépatomes [13]. Des récepteurs de la même famille (CXCR4, bradykinine B2, apelin), ainsi que de nombreux récepteurs à activité tyrosine-kinase de facteurs de croissance (epidermal growth factor - EGF, nerve growth factor - NGF, fibroblast growth factor - FGF), présentent également une localisation nucléaire : un mode d’action intracrine par lequel le ligand intracellulaire activerait son récepteur nucléaire fonctionnel [13, 14] pourrait participer à l’activité mitogène du récepteur.
Outre les cellules tumorales, l’endothélium des vaisseaux intratumoraux exprime également ETA-R, ETB-R et/ou AT1R, qui participent à l’angiogenèse associée à la progression tumorale (voir plus loin). Enfin, bien que les relations entre la cellule tumorale et son micro-environnement soient encore mal connues, deux publications récentes font état de l’expression d’AT1R par des macrophages associés à la tumeur et par des cellules stromales, dans des modèles murins de carcinogenèse [15, 16].
Mécanismes d’action de l’endothéline-1 et de l’angiotensine II sur la croissance tumorale et l’angiogenèse
ET-1, AngII et croissance tumorale
ET-1 et AngII ont une action mitogène ou anti-apoptotique sur de nombreux types cellulaires (ovaire, peau, prostate, sein, côlon et système nerveux central) [5-7, 9, 10, 12, 17]. Selon le type cellulaire, l’un ou l’autre des deux sous-types de récepteur d’ET-1 peut être impliqué dans ces réponses, alors qu’AngII agit toujours par l’intermédiaire d’AT1R. Comme d’autres récepteurs mitogènes, ils induisent l’activation de la voie ERK, principalement par transactivation du récepteur de l’EGF (EGFR) (Figure 2), dont le rôle sur la croissance tumorale est bien documenté [18]. D’autres voies de signalisation, parmi lesquelles les voies PI3K/Akt et FAK, sont également impliquées dans les effets de ces récepteurs sur la prolifération, la survie et la migration cellulaires (Figure 2).
Figure 2
Principales voies de signalisation intracellulaire, couplées aux récepteurs de l’endothéline 1 (ET-1) et de l’angiotensine II (AngII), impliquées dans la progression tumorale.

Les récepteurs d’ET-1 (ETA-R et ETB-R) et le récepteur AT1R de l’AngII sont couplés positivement aux voies ERK (extracellular signal-regulated kinase), PI3K (phospsho-inositide 3 kinase)/Akt, et FAK (focal adhesion kinase), tandis qu’AT2R y est couplé négativement. Un des mécanismes d’action est la trans-activation du récepteur de l’EGF (epidermal growth factor) (EGFR) via le clivage du ligand HB-EGF (heparin binding EGF-like growth factor) par une métalloprotéase (pour les récepteurs ETA-R et ETB-R, AT1R), ou la trans-inhibition d’EGFR, impliquant probablement ATIP (AT2 receptor-interacting protein) et SHP-1 (Src homology region 2 domain-containing phosphatase 1) (pour le récepteur AT2R). ET-1 comme AngII sont capables d’induire la production de VEGF (vascular endothelium growth factor) en augmentant le niveau d’expression d’HIF1α (hypoxia-induced factor)
En accord avec son action antiproliférative et pro-apoptotique, AT2R trans-inactive l’EGFR [19] (Figure 2), notamment via l’activation de la tyrosine phosphatase SHP-1 (Src homology region 2 domain-containing phosphatase 1) [3], dont le rôle dans le cancer a été rapporté. Une nouvelle famille de protéines interagissant avec AT2R (ATIP), capables d’inhiber la prolifération cellulaire induite par l’EGF, a été identifiée [20], et des études récentes suggèrent un rôle potentiel d’ATIP en tant que suppresseur de tumeur.
ET-1, AngII et angiogenèse
Parallèlement aux facteurs angiogéniques VEGF (vascular endothelium growth factor), FGF ou HGF (hepatocyte growth factor), ET-1 et AngII apparaissent désormais aussi comme des acteurs de l’angiogenèse (pour revues, voir [21, 22]). Ces deux peptides, en agissant directement sur les cellules endothéliales par l’intermédiaire d’ETB-R et d’AT1R, sont capables de moduler in vitro les étapes précoces de l’angiogenèse (prolifération, migration, invasion, production de métalloprotéases spécifiques de la matrice extracellulaire - MMP), mais aussi l’étape, plus tardive, de tubulogenèse. ET-1 et AngII, en activant respectivement ETA-R et AT1R, sont également de puissants mitogènes pour les cellules musculaires lisses vasculaires. Au contraire, AT2R, par son action négative sur les voies de signalisation couplées à AT1R et au récepteur du VEGF, régule négativement la migration des cellules endothéliales et la tubulogenèse.
Il faut en outre noter qu’ET-1 et AngII stimulent indirectement l’angiogenèse tumorale via la production de VEGF, aussi bien par les cellules tumorales que par les cellules endothéliales. Cela implique notamment l’induction d’HIF-1α, le facteur de réponse à l’hypoxie (hypoxia-induced factor) contrôlant la production de VEGF (Figure 2). Une augmentation de la sécrétion de VEGF par les macrophages associés à la tumeur et par les cellules stromales a également été observée en réponse à l’AngII [15, 16]. Réciproquement, le VEGF induit la sécrétion d’ET-1 par les cellules endothéliales et les cellules musculaires lisses, conduisant à une boucle d’amplification de la production de ces facteurs pro-angiogéniques. Finalement, ET-1 comme AngII sont sécrétés en réponse à plusieurs stimuli associés au processus tumoral (hypoxie, cytokines inflammatoires, facteurs de croissance), et peuvent agir de concert avec le VEGF sur les différentes étapes de l’angiogenèse.
Approches thérapeutiques
Les systèmes ET-1/ETA-R et AngII/AT1R sont depuis plusieurs années des cibles thérapeutiques dans le domaine de l’hypertension, en liaison avec leur activité vasoconstrictrice. L’utilisation d’antagonistes des récepteurs ETA-R ou AT1R, ou d’inhibiteurs de l’ECA (iECA), fait désormais l’objet de protocoles cliniques établis. Plus récemment, ces mêmes antagonistes ou inhibiteurs ont montré une activité antitumorale dans des modèles précliniques (pour les deux systèmes) et dans plusieurs essais cliniques (pour le système ET-1).
Inhibition des systèmes ET-1 et Ang II dans des modèles animaux
Plusieurs modèles in vivo ont démontré le rôle d’antagonistes spécifiques d’ETA-R, ETB-R ou AT1R, ainsi que d’inhibiteurs de l’ECA (iECA), dans différentes étapes de la carcinogenèse : croissance tumorale, angiogenèse et formation de métastases (Tableau II).
Tableau II
Traitements antitumoraux in vivo par des antagonistes des récepteurs de l’endothéline 1 et de l’angiotensine II, et par des inhibiteurs de synthèse de l’angiotensine II.
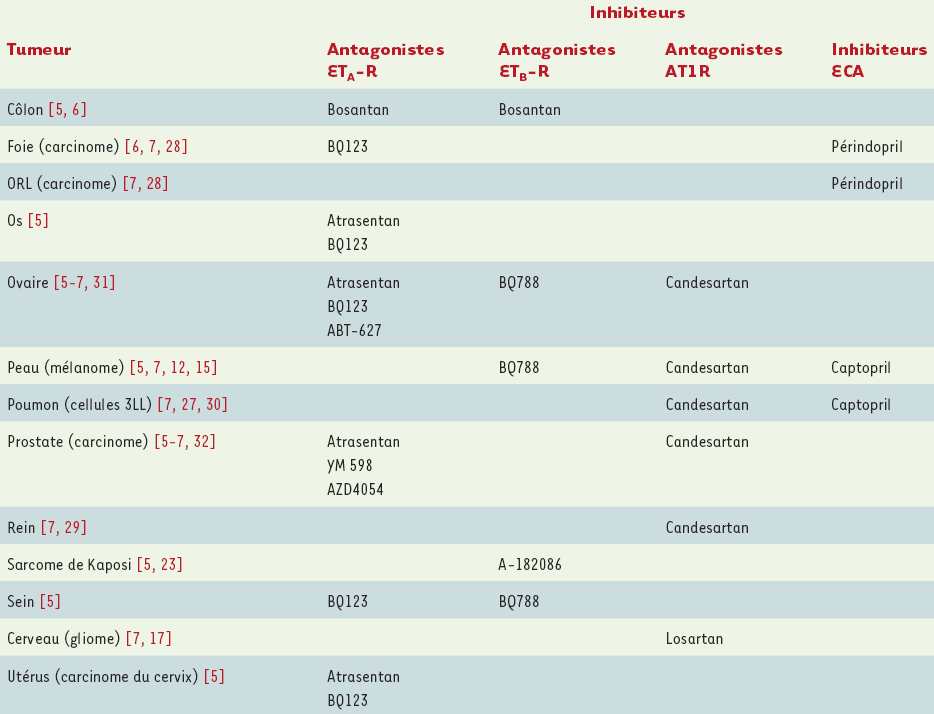
L’effet bénéfique d’antagonistes des récepteurs d’ET-1 sur le développement de diverses tumeurs [5, 6, 12, 23, 24] a ainsi été montré dans des modèles syngéniques : un antagoniste d’ETA-R (BQ123) permet de réduire la croissance de tumeurs hépatiques, tandis qu’un antagoniste non sélectif des récepteurs d’ET-1 (bosantan) ralentit l’évolution d’adénocarcinomes du colon vers un grade plus élevé. Dans des modèles de souris athymiques, une diminution de la croissance de tumeurs humaines implantées a été obtenue à l’aide d’antagonistes spécifiques du sous-type de récepteur exprimé par la tumeur : ETA-R (A127722) pour les tumeurs osseuses, ETB-R (BQ788) pour les mélanomes. De plus, le blocage des récepteurs d’ET-1 peut conduire à une inhibition de l’angiogenèse, comme cela a été mis en évidence, avec un antagoniste d’ETA-R (ABT-627), pour des carcinomes ovariens dans un modèle de xénogreffe. Dans un modèle de xénogreffe de cellules de sarcome de Kaposi, l’invasion et la croissance tumorales sont également ralenties par l’utilisation d’un antagoniste non sélectif des récepteurs d’ET-1 (A-182086), du fait de l’inhibition de la production de MMP-2 et MMP-9 [23]. Les cancers du sein et de la prostate à un stade avancé produisent souvent des métastases osseuses dont la formation est dépendante d’ET-1 [24, 25] : un rôle inhibiteur de l’antagoniste d’ETA-R (ABT-627) a été mis en évidence dans des modèles expérimentaux [24].
Pour le système AngII, les premières observations montrant les effets anti-angiogéniques et antiprolifératifs de l’iECA captopril dans un modèle syngénique de fibrosarcome de rat [26] ont par la suite été étendues à divers types de tumeurs. En effet, l’utilisation du captopril permet d’atténuer la croissance tumorale et l’angiogenèse du carcinome pulmonaire de Lewis (3LL) chez la souris [27], et de réduire de façon significative la taille des tumeurs dans des modèles de xénogreffe de carcinomes rénaux et de mélanomes humains [7]. De plus, le captopril permet de diminuer le nombre de métastases pulmonaires et d’augmenter la survie des souris, en inhibant l’expression et l’activité des MMP-2 et MMP-9 [26, 27]. Il est intéressant de noter que le captopril bloque l’activité de l’ECA, et donc la synthèse d’AngII, mais peut également avoir une action inhibitrice directe, indépendante de l’AngII, sur le site actif des MMP-2 et MMP-9 [26, 27]. L’utilisation d’un autre iECA, le périndopril, permet également de réduire significativement la croissance et la vascularisation tumorales dans des modèles murins de carcinomes hépatiques et des voies ORL. Seul ou en combinaison avec d’autres traitements, il réduit l’angiogenèse tumorale en inhibant l’expression du VEGF, la migration des cellules endothéliales et leur formation tubulaire [28]. L’utilisation plus récente d’antagonistes spécifiques d’AT1R (candesartan, losartan) a également donné des résultats intéressants, avec une réduction de la croissance et de la vascularisation des tumeurs dans des modèles syngéniques de cancer rénal [29], de sarcome (S-180) [30], de mélanome (B16-F1) [15] et de gliome (C6) [17], ainsi que dans des modèles xénogéniques de cancers de la prostate et de l’ovaire [7, 31]. Le candesartan s’est également révélé actif pour réduire le nombre de métastases pulmonaires provenant de carcinomes de Lewis (3LL) et de carcinomes rénaux [29, 30].
Inhibition du système ET-1 dans des essais cliniques
Des essais cliniques impliquant l’inhibition du système ET-1 ont récemment été menés dans le contexte de thérapies antitumorales.
Le traitement du cancer de la prostate par administration orale d’un antagoniste d’ETA-R, l’atrasentan (actuellement en phase clinique III), constitue ainsi un véritable progrès [32, 5]. Une approche similaire, utilisant l’atrasentan, donne également de premiers résultats encourageants dans le cadre du traitement d’adénocarcinomes [33]. Les effets secondaires liés aux conséquences physiologiques du blocage d’ETA-R par des antagonistes (maux de tête, hypotension, oedème périphérique) sont généralement limités, et bien tolér és. Ces premières données suggèrent que l’utilisation d’antagonistes spécifiques de sous-types de récepteur d’ET-1 pourrait être élargie aux différentes tumeurs exprimant ETA-R ou ETB-R. De plus, on peut supposer que l’action inhibitrice de ces antagonistes sur l’angiogenèse pourrait également contribuer à ralentir la progression tumorale.
De façon analogue à celle du système ET-1, l’inhibition du système AngII pourrait avoir des applications thérapeutiques antitumorales. L’observation d’effets bénéfiques d’un traitement à long terme par un iECA, inhibiteur de la synthèse d’AngII, dans des cas de cancers du sein ou du poumon [34], reste cependant sujette à controverse [35] ; ces résultats doivent être confirmés, de même que doit être testée l’utilisation d’antagonistes spécifiques d’AT1R pour le blocage du système AngII.
Conclusions et perspectives
De manière remarquable, ET-1 et AngII, deux peptides connus pour leur activité vasoactive, présentent également une activité mitogène et pro-angiogénique par l’intermédiaire de leurs récepteurs, qui appartiennent à la même superfamille des RCPG. Outre leur expression commune sur l’endothélium vasculaire, les récepteurs des systèmes ET-1 et AngII présentent un profil d’expression relativement large, et peuvent être impliqués dans le développement tumoral de différents tissus ou types cellulaires (Tableau I). Il est intéressant de souligner qu’ET-1 et AngII régulent réciproquement leur sécrétion, et peuvent agir de concert sur diverses réponses cellulaires, dont l’angiogenèse.
L’effet opposé des deux sous-types de récepteurs d’AngII sur la réponse mitogène suggère qu’une thérapie bloquant spécifiquement AT1R pourrait être préférable à une thérapie inhibant la synthèse d’AngII. En revanche, l’utilisation d’antagonistes mixtes pour ETA-R et ETB-R pourrait permettre de cibler à la fois la croissance et l’angiogenèse tumorales. De fait, le blocage du système ET-1 commence à donner des résultats encourageants en monothérapie antitumorale. Au vu des données présentées dans cette revue, il est tentant de spéculer qu’une thérapie combinée ciblant les deux systèmes ET-1 et AngII, à l’image de certaines stratégies thérapeutiques récentes de l’hypertension et de pathologies cardiaques et rénales, pourrait être bénéfique pour le traitement de tumeurs fortement angiogéniques.
Parties annexes
Remerciements
Cette étude a bénéficié du soutien financier du Centre national de la Recherche scientifique (CNRS), de l’Institut national pour la Santé et la Recherche médicale (Inserm), de l’Université Paris V, et de subventions accordées par l’Association pour la Recherche sur le Cancer (ARC), la Ligue nationale contre le Cancer Comité Ile-de-France (LNCC) et la Fondation pour la Recherche médicale (FRM).
Références
- 1. Lotersztajn S. Les endothélines. Med Sci (Paris) 1993 ; 9 : 1084-93.
- 2. Cazaubon S, Couraud PO. Nitric oxide and endothelin at the blood-brain barrier. In : Pardrige W, ed. Introduction to the blood-brain barrier. New York Raven Press Ltd, 1998 : 338-44.
- 3. Nouet S, Nahmias C. Signal transduction from the angiotensin II AT2 receptor. Trends Endocrinol Metab 2000 ; 11 : 1-6.
- 4. Nelson JB, Hedican SP, George DJ, et al. Identification of endothelin-1 in the pathophysiology of metastatic adenocarcinoma of the prostate. Nat Med 1995 ; 1 : 944-9.
- 5. Nelson J, Bagnato A, Battistini B, Nisen P. The endothelin axis : emerging role in cancer. Nat Rev Cancer 2003 ; 3 : 110-6.
- 6. Grant K, Loizidou M, Taylor I. Endothelin-1 : a multifunctional molecule in cancer. Br J Cancer 2003 ; 88 : 163-6.
- 7. Deshayes F, Nahmias C. Angiotensin II receptors : a new role in cancer ? Trends Endocrinol Metab 2005 ; 16 : 293-9.
- 8. Egidy G, Baviera E, Ciuffo G, et al. Localization of the endothelin system in aldosterone-producing adenomas. Hypertension 2001 ; 38 : 1137-42.
- 9. Juillerat-Jeanneret L, Celerier J, Chapuis Bernasconi C, et al. Renin and angiotensinogen expression and functions in growth and apoptosis of human glioblastoma. Br J Cancer 2004 ; 90 : 1059-68.
- 10. Anguelova E, Beuvon F, Leonard N, et al. Functional endothelin ET(B) receptors are selectively expressed in human oligodendrogliomas. Brain Res Mol Brain Res 2005 ; 137 : 77-88.
- 11. Lazarini F, Strosberg AD, Couraud PO, Cazaubon SM. Coupling of ETB endothelin receptor to mitogen-activated protein kinase stimulation and DNA synthesis in primary cultures of rat astrocytes. J Neurochem 1996 ; 66 : 459-65.
- 12. Lahav R, Heffner G, Patterson PH. An endothelin receptor B antagonist inhibits growth and induces cell death in human melanoma cells in vitro and in vivo. Proc Natl Acad Sci USA 1999 ; 96 : 11496-500.
- 13. Cook JL, Zhang Z, Re RN. In vitro evidence for an intracellular site of angiotensin action. Circ Res 2001 ; 89 : 1138-46.
- 14. Boivin B, Chevalier D, Villeneuve LR, et al. Functional endothelin receptors are present on nuclei in cardiac ventricular myocytes. J Biol Chem 2003 ; 278 : 29153-63.
- 15. Egami K, Murohara T, Shimada T, et al. Role of host angiotensin II type 1 receptor in tumor angiogenesis and growth. J Clin Invest 2003 ; 112 : 67-75.
- 16. Fujita M, Hayashi I, Yamashina S, et al. Angiotensin type 1a receptor signaling-dependent induction of vascular endothelial growth factor in stroma is relevant to tumor-associated angiogenesis and tumor growth. Carcinogenesis 2005 ; 26 : 271-9.
- 17. Arrieta O, Guevara P, Escobar E, et al. Blockage of angiotensin II type I receptor decreases the synthesis of growth factors and induces apoptosis in C6 cultured cells and C6 rat glioma. Br J Cancer 2005 ; 92 : 1247-52.
- 18. Fischer O, Hart S, Gschwind A, Ullrich A. EGFR signal transactivation in cancer cells. Biochem Soc Trans 2003 ; 31 : 1203-8.
- 19. Elbaz N, Bedecs K, Masson M, et al. Functional trans-inactivation of insulin receptor kinase by growth-inhibitory angiotensin II AT2 receptor. Mol Endocrinol 2000 ; 14 : 795-804.
- 20. Nouet S, Amzallag N, Li J, et al. Trans-inactivation of receptor tyrosine kinases by novel angiotensin II AT2 receptor-interacting protein, ATIP. J Biol Chem 2004 ; 279 : 28989-97.
- 21. Bagnato A, Spinella F. Emerging role of endothelin-1 in tumor angiogenesis. Trends Endocrinol Metab 2003 ; 14 : 44-50.
- 22. Escobar E, Rodriguez-Reyna TS, Arrieta O, Sotelo J. Angiotensin II, cell proliferation and angiogenesis regulator : biologic and therapeutic implications in cancer. Curr Vasc Pharmacol 2004 ; 2 : 385-99.
- 23. Rosano L, Spinella F, Di Castro V, et al. Endothelin receptor blockade inhibits molecular effectors of Kaposi’s sarcoma cell invasion and tumor growth in vivo. Am J Pathol 2003 ; 163 : 753-62.
- 24. Yin JJ, Mohammad KS, Kakonen SM, et al. A causal role for endothelin-1 in the pathogenesis of osteoblastic bone metastases. Proc Natl Acad Sci USA 2003 ; 100 : 10954-9.
- 25. Chiao JW, Moonga BS, Yang YM, et al. Endothelin-1 from prostate cancer cells is enhanced by bone contact which blocks osteoclastic bone resorption. Br J Cancer 2000 ; 83 : 360-5.
- 26. Volpert OV, Ward WF, Lingen MW, et al. Captopril inhibits angiogenesis and slows the growth of experimental tumors in rats. J Clin Invest 1996 ; 98 : 671-9.
- 27. Prontera C, Mariani B, Rossi C, et al. Inhibition of gelatinase A (MMP-2) by batimastat and captopril reduces tumor growth and lung metastases in mice bearing Lewis lung carcinoma. Int J Cancer 1999 ; 81 : 761-6.
- 28. Lindberg H, Nielsen D, Jensen B, et al. Angiotensin converting enzyme inhibitors for cancer treatment ? Acta Oncol 2004 ; 43 : 142-52.
- 29. Miyajima A, Kosaka T, Asano T, et al. Angiotensin II type I antagonist prevents pulmonary metastasis of murine renal cancer by inhibiting tumor angiogenesis. Cancer Res 2002 ; 62 : 4176-9.
- 30. Fujita M, Hayashi I, Yamashina S, et al. Blockade of angiotensin AT1a receptor signaling reduces tumor growth, angiogenesis, and metastasis. Biochem Biophys Res Commun 2002 ; 294 : 441-7.
- 31. Suganuma T, Ino K, Shibata K, et al. Functional expression of the angiotensin II type 1 receptor in human ovarian carcinoma cells and its blockade therapy resulting in suppression of tumor invasion, angiogenesis, and peritoneal dissemination. Clin Cancer Res 2005 ; 11 : 2686-94.
- 32. Carducci MA, Padley RJ, Breul J, et al. Effect of endothelin-A receptor blockade with atrasentan on tumor progression in men with hormone-refractory prostate cancer : a randomized, phase II, placebo-controlled trial. J Clin Oncol 2003 ; 21 : 679-89.
- 33. Carducci MA, Nelson JB, Bowling MK, et al. Atrasentan, an endothelin-receptor antagonist for refractory adenocarcinomas : safety and pharmacokinetics. J Clin Oncol 2002 ; 20 : 2171-80.
- 34. Lever A, Hole D, Gillis C, et al. Do inhibitors of angiotensin-I-converting enzyme protect against risk of cancer ? Lancet 1998 ; 352 : 179-84.
- 35. Friis S, Sorensen HT, Mellemkjaer L, et al. Angiotensin-converting enzyme inhibitors and the risk of cancer : a population-based cohort study in Denmark. Cancer 2001 ; 92 : 2462-70.
Liste des figures
Figure 1
Similitudes entre les étapes de synthèse de l’endothéline 1 (ET-1) et de l’angiotensine II (AngII) et leurs modes d’action.
ET-1 et AngII sont tous deux des produits de clivage de précurseurs inactifs par une métalloprotéase membranaire, respectivement l’ECE-1 (enzyme de conversion d’ET-1) et l’ECA (enzyme de conversion de l’angiotensine I). Ils agissent par l’intermédiaire de récepteurs appartenant à la même superfamille des récepteurs couplés aux protéines G (RCPG) : ETA-R et ETB-R pour ET-1, AT1R et AT2R pour AngII. Alors qu’ETA-R, ETB-R et AT1R sont couplés positivement à la prolifération, AT2R a une action antiproliférative. L’endothélium vasculaire est une des cibles communes d’ET-1 et AngII.
Figure 2
Principales voies de signalisation intracellulaire, couplées aux récepteurs de l’endothéline 1 (ET-1) et de l’angiotensine II (AngII), impliquées dans la progression tumorale.
Les récepteurs d’ET-1 (ETA-R et ETB-R) et le récepteur AT1R de l’AngII sont couplés positivement aux voies ERK (extracellular signal-regulated kinase), PI3K (phospsho-inositide 3 kinase)/Akt, et FAK (focal adhesion kinase), tandis qu’AT2R y est couplé négativement. Un des mécanismes d’action est la trans-activation du récepteur de l’EGF (epidermal growth factor) (EGFR) via le clivage du ligand HB-EGF (heparin binding EGF-like growth factor) par une métalloprotéase (pour les récepteurs ETA-R et ETB-R, AT1R), ou la trans-inhibition d’EGFR, impliquant probablement ATIP (AT2 receptor-interacting protein) et SHP-1 (Src homology region 2 domain-containing phosphatase 1) (pour le récepteur AT2R). ET-1 comme AngII sont capables d’induire la production de VEGF (vascular endothelium growth factor) en augmentant le niveau d’expression d’HIF1α (hypoxia-induced factor)
Liste des tableaux
Tableau I
Expression des récepteurs de l’endothéline 1 et de l’angiotensine II dans différents tissus tumoraux et lignées cellulaires tumorales.
+ : exprimé ; ++ : surexprimé ; - : non exprimé ; +/- : faiblement détecté. CDIS : carcinome ductal in situ.
Tableau II
Traitements antitumoraux in vivo par des antagonistes des récepteurs de l’endothéline 1 et de l’angiotensine II, et par des inhibiteurs de synthèse de l’angiotensine II.