Corps de l’article
Le maintien du taux de glucose sanguin dans des valeurs étroites est une nécessité physiologique comme le soulignent les complications sévères liées au diabète. L’homéostasie glucidique résulte de l’action conjointe de plusieurs organes qui équilibrent leurs capacités à produire ou à utiliser du glucose. Parmi ces organes, le foie joue un rôle important dans cet équilibre car il représente une source essentielle de glucose au cours du jeûne. Afin de réduire la glycémie excessive des patients diabétiques de type 2, la metformine (Glucophage®), une molécule de la famille des biguanides, est utilisée couramment depuis plusieurs dizaines d’années sans que l’on connaisse encore aujourd’hui son mécanisme d’action cellulaire exact. La mise en évidence par Shaw et al. [1] du rôle du suppresseur de tumeur LKB1 (encore appelé STK11 pour sérine thréonine protéine kinase 11) dans le mode d’action de la metformine, ouvre désormais de nouvelles perspectives dans le traitement du diabète mais également celui du cancer. LKB1 est une protéine kinase dont des mutations ont été identifiées comme étant à l’origine du syndrome de Peutz-Jeghers, maladie héréditaire rare caractérisée par le développement de polypes intestinaux et d’adénocarcinomes pulmonaires. La première cible de phosphorylation de LKB1 à avoir été identifiée est la protéine kinase activée par l’AMP ou AMPK (AMP-activated protein kinase), un senseur métabolique qui permet d’ajuster, en permanence et au plus près, les besoins et disponibilité énergétiques de la cellule [2, 3]. Parmi les hypothèses permettant d’expliquer la formation de tumeurs chez les patients présentant une mutation de LKB1, des défauts de signalisation dans les voies impliquant le complexe TSC/mTOR ou la protéine p53, qui sont connues pour être contrôlées par l’AMPK [4, 5], ont été avancés. L’AMPK semble toutefois exercer un rôle majeur dans le contrôle de la production hépatique de glucose puisqu’il a été montré que son activation inhibe fortement l’expression des gènes clés de la gluconéogenèse que sont la glucose 6-phosphatase (G6Pase) et la phosphoénolpyruvate carboxykinase (PEPCK) [6]. Dans l’élucidation des mécanismes d’action cellulaire de la metformine, des avancées récentes ont permis de mettre en évidence un rôle potentiel de l’activation de l’AMPK par cet agent antidiabétique dans le contrôle de la production hépatique de glucose [7]. De même, une amélioration de l’homéostasie glucidique et de la sensibilité à l’insuline de modèles murins diabétiques traités avec des activateurs de l’AMPK a également été démontrée [2]. Cependant, la découverte récente d’activités AMPK kinase autres que LKB1 a posé la question du rôle de LKB1 dans les effets de l’AMPK en relation avec le contrôle de l’homéostasie glucidique. Pour répondre à cette interrogation, une délétion hépatique de LKB1 a été réalisée dans le foie d’animaux adultes par l’infection d’animaux LKB1 floxés avec un adénovirus exprimant la recombinase Cre [1]. Cette délétion hépatique de LKB1 induit une perte de la phosphorylation de l’AMPK et provoque une augmentation importante de la glycémie ainsi qu’une intolérance au glucose (incapacité à normaliser la glycémie en réponse à une charge en glucose). Ces animaux ne présentent cependant pas de résistance apparente à l’action de l’insuline, ce qui indique que l’activation du transport de glucose dans les tissus périphériques n’est pas altérée. L’hyperglycémie des animaux délétés en LKB1 hépatique est vraisemblablement due à une production hépatique de glucose qui n’est plus contrôlée chez ces animaux, en lien direct avec l’augmentation de l’expression des enzymes de la gluconéogenèse, notamment la G6Pase et la PEPCK.
Au cours du jeûne, l’initiation du programme gluconéogénique repose conjointement sur l’activation du facteur de transcription CREB (CRE-binding protein) par la PKA et le recrutement du coactivateur, TORC2 (transducer of regulated CREB activity 2) [8] (Figure 1). L’activité de TORC2 est contrôlée par phosphorylation par l’AMPK sur le résidu Ser171, déterminant sa liaison à la protéine 14-3-3 et sa localisation subcellulaire [8, 9] (Figure 2). Dans le foie de souris à jeun, TORC2 est déphosphorylé et transloqué dans le noyau où il augmente l’expression du coactivateur PGC-1α (peroxisome proliferator-activated receptor gamma coactivator-1α), impliqué dans l’activation transcriptionnelle des gènes G6Pase et PEPCK en association avec les facteurs de transcription HNF4α et Foxo1 (Figure 1). À l’inverse, la déplétion de TORC2 dans les hépatocytes provoque une diminution importante de l’expression de la PEPCK, et donc de la glycémie au cours du jeûne [8]. L’apport récent des travaux de Shaw et al. montre, qu’en l’absence de LKB1, TORC2 n’est plus phosphorylé et est présent majoritairement dans le noyau des hépatocytes [1]. Pour démontrer le rôle de cette translocation nucléaire de TORC2 dans l’augmentation de la gluconéogenèse chez les animaux délétés en LKB1 dans le foie, les niveaux d’expression de TORC2 hépatique ont été artificiellement réduits par l’injection d’un adénovirus exprimant un ARN interférent. Dans le foie de ces animaux dont la glycémie est initialement emballée, la diminution de PGC-1α est proportionnelle à celle de TORC2, ce qui conduit, au final, à une diminution du glucose sanguin à jeun [1]. Ces données indiquent que le coactivateur TORC2 est une cible de LKB1 et qu’il joue un rôle crucial dans le contrôle de la gluconéogenèse. Enfin, l’originalité de cette étude a été d’établir le rôle de LKB1 dans l’inhibition de la gluconéogenèse hépatique induite par la metformine. En effet, l’administration in vivo de metformine chez les animaux délétés en LKB1 hépatique n’a aucun effet sur la glycémie et l’activation de l’AMPK hépatique, contrairement aux animaux témoins [1], suggérant l’importance du système LKB1/AMPK dans le mode d’action de cet antidiabétique. De plus, ces résultats permettent de démontrer que l’effet normoglycémiant de la metformine, dont l’origine était jusque-là très discutée, est principalement hépatique via une inhibition de la gluconéogenèse. La description de LKB1 comme suppresseur de tumeur questionne aujourd’hui sur une action antitumorale possible de la metformine et il serait intéressant de mesurer l’incidence des cancers chez les patients diabétiques sous traitement avec la metformine [10]. En conclusion, ces travaux renforcent l’idée de l’existence d’interfaces communes dans le traitement du diabète et du cancer, qui restent cependant encore à explorer.
Figure 1
Régulation transcriptionnelle des gènes de la gluconéogenèse au cours du jeûne.
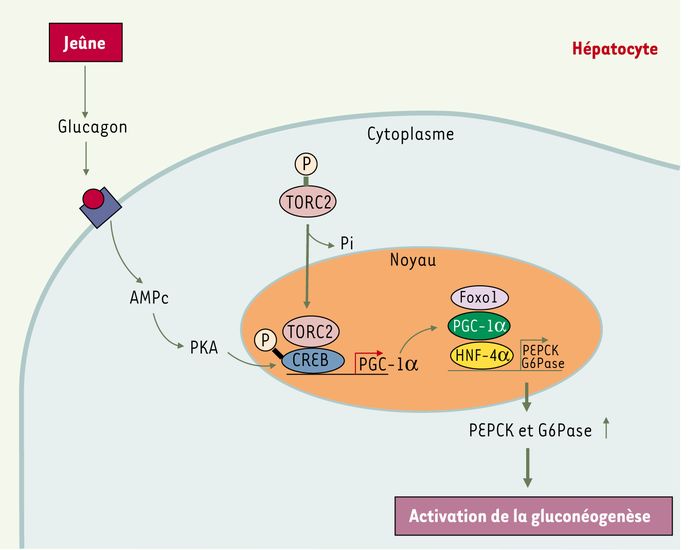
En réponse à l’activation par la PKA, le facteur de transcription CREB recrute le coactivateur TORC2 et permet l’expression du gène codant le coactivateur PGC-1α. En association avec les facteurs de transcription HNF4α et Foxo1, PGC-1α active la transcription des gènes G6Pase et PEPCK provoquant une activation de la gluconéogenèse hépatique.
Figure 2
Mode d’action de la metformine dans l’inhibition de la gluconéogenèse hépatique.

En réponse à l’action de la metformine et à l’activation par le suppresseur de tumeur LKB1, l’AMPK phosphoryle le coactivateur TORC2 sur le résidu Ser171 et inhibe sa translocation dans le noyau. La séquestration de TORC2 dans le cytoplasme par la protéine 14-3-3 empêche l’activation du programme gluconéogénique et inhibe la production hépatique de glucose
Parties annexes
Références
- 1. Shaw RJ, Lamia KA, Vasquez D, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 2005 ; 310 : 1642-6.
- 2. Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase : ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab 2005 ; 1 : 15-25.
- 3. Foretz M, Taleux N, Guigas B, et al. Régulation du métabolisme énergétique par l’AMPK : une nouvelle voie thérapeutique pour le traitement des maladies métaboliques et cardiaques. Med Sci (Paris) 2006 ; 22 (sous presse).
- 4. Jones RG, Plas DR, Kubek S, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell 2005 ; 18 : 283-93.
- 5. Shaw RJ, Bardeesy N, Manning BD, et al. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell 2004 ; 6 : 91-9.
- 6. Foretz M, Ancellin N, Andreelli F, et al. Short-term overexpression of a constitutively active form of AMP-activated protein kinase in the liver leads to mild hypoglycemia and fatty liver. Diabetes 2005 ; 54 : 1331-9.
- 7. Zhou G, Myers R, Li Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 2001 ; 108 : 1167-74.
- 8. Koo SH, Flechner L, Qi L, et al. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature 2005 ; 437 : 1109-11.
- 9. Screaton RA, Conkright MD, Katoh Y, et al. The CREB coactivator TORC2 functions as a calcium- and cAMP-sensitive coincidence detector. Cell 2004 ; 119 : 61-74.
- 10. Evans JM, Donnelly LA, Emslie-Smith AM, et al. Metformin and reduced risk of cancer in diabetic patients. Br Med J 2005 ; 330 : 1304-5.
Liste des figures
Figure 1
Régulation transcriptionnelle des gènes de la gluconéogenèse au cours du jeûne.
En réponse à l’activation par la PKA, le facteur de transcription CREB recrute le coactivateur TORC2 et permet l’expression du gène codant le coactivateur PGC-1α. En association avec les facteurs de transcription HNF4α et Foxo1, PGC-1α active la transcription des gènes G6Pase et PEPCK provoquant une activation de la gluconéogenèse hépatique.
Figure 2
Mode d’action de la metformine dans l’inhibition de la gluconéogenèse hépatique.
En réponse à l’action de la metformine et à l’activation par le suppresseur de tumeur LKB1, l’AMPK phosphoryle le coactivateur TORC2 sur le résidu Ser171 et inhibe sa translocation dans le noyau. La séquestration de TORC2 dans le cytoplasme par la protéine 14-3-3 empêche l’activation du programme gluconéogénique et inhibe la production hépatique de glucose