Corps de l’article
Il est communément admis que la migraine présente une forte composante génétique. À cet égard, des travaux récents qui ont établi que des formes autosomiques rares de migraine avec aura, migraine hémiplégique familiale (FHM), sont associées à des mutations faux-sens dans au moins deux gènes, ont ouvert de nouvelles perspectives sur la physiologie de la migraine. Ces deux gènes, CACNA1A qui code la sous-unité α1 du canal neuronal de Ca2+ sensible au voltage (CaV2.1) et ATP1A2 qui code l’isoforme α2 de la Na,K-ATPase sont impliqués dans la FHM1 et la FHM2 respectivement. À eux deux, ils répondent à la plupart des cas de FHM [1]. Récemment néanmoins, une mutation faux-sens (G1489K) dans le gène SCN1A du canal ionique du Na+ dépendant du voltage neuronal a également été signalée [2]. Il est probable que, considérées conjointement, la FHM et peut-être d’autres formes de migraine résultent d’une déficience du transport normal de cations. Cet article discute la nature des altérations fonctionnelles susceptibles de justifier le phénotype de la FHM2.
La structure de la Na,K-ATPase, diversité de fonctions et d’isoformes
La Na,K-ATPase catalyse l’échange électrogène, réglé par l’ATP, de trois ions Na+CYT contre deux ions K+EXT à travers la membrane plasmique de presque toutes les cellules animales ; elle est essentielle à l’entretien des gradients électrochimiques de cations alcalins qui sont abolis par les canaux ioniques dans la propagation des potentiels d’action [3, 4]. Cette pompe comprend une grande (˜110 kDa) sous-unité catalytique α qui possède 10 segments transmembranaires et une sous-unité β plus petite, hautement glycosylée, qui assure le repliement et l’ancrage appropriés de la sous-unité α dans la membrane. Quatre isoformes de α et trois de β ont été décrites jusqu’ici ; elles sont distribuées de manière tissu- et développement-dépendante. Chez les mammifères adultes, α2 se trouve principalement dans le muscle squelettique et le cerveau, en particulier dans les cellules gliales, et dans une moindre mesure dans le coeur, les adipocytes et l’oeil. La Figure 1 illustre un cycle de réaction abrégé de la Na,K-ATPase qui, comme toutes les pompes de type P, implique des transitions de conformation des formes déphospho- et phosphorylées de l’enzyme.
Figure 1
Le cycle de réaction de la Na,K-ATPase.
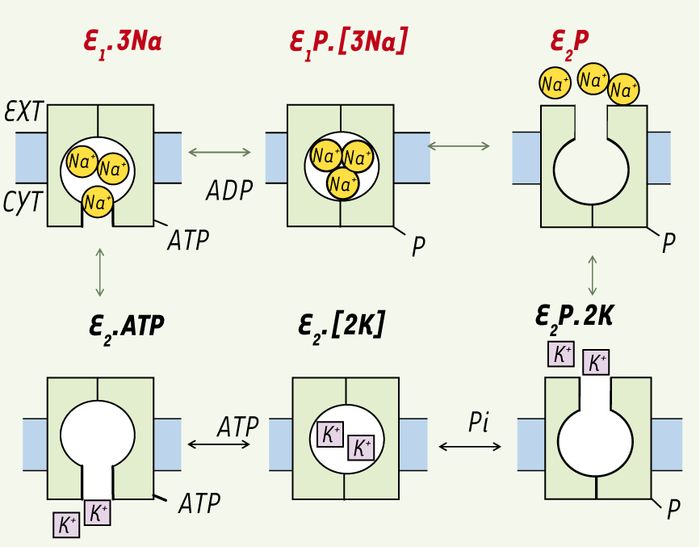
Selon le modèle « Albers-Post » abrégé, l’enzyme en E1 lie le Na+CYT et l’ATP avec haute affinité. L’enzyme est alors phosphorylé sur un résidu aspartate dans la boucle cytoplasmique TM4-TM5 (voir Figure 2) menant à l’occlusion des ions de Na+ (E1P.[3Na]). Par suite d’un changement de conformation en E2P, l’affinité apparente pour le K+EXT est nettement augmentée et diminuée pour le Na+ tel que ces derniers sont relâchés en faveur de deux K+EXT (E2P.2K). Les ions de K+ sont alors occlus en E2P.[2K] qui est rapidement déphosphorylée pour donner E2.[2K]. Les ions de K+ sont à leur tour relâchés, l’enzyme lie l’ATP (liaison à basse affinité) pour former E2.ATP qui change rapidement de conformation de sorte que l’enzyme est prêt pour un autre cycle. Le cycle catalytique de cette pompe ionique de type P implique une translocation de Na+ et K+ couplée à une hydrolyse ATP impliquant une phosphorylation et une déphosphorylation de la sous-unité catalytique α (résidu aspartyl sur l’enzyme, E) et des transitions conformationnelles de la phospho- et déphosphoenzyme (E1P→E2P et E1→E2). Ext : extracellulaire ; Cyt : cytoplasmique.
FHM2 associée à des mutations faux-sens dans l’isoforme a2 de la Na,K-ATPase
Jusqu’ici, plus de 20 allèles du gène α2 ont été relevés chez les membres de familles où sévit la FHM2 (Figure 2). La plupart des allèles sont des mutations faux-sens et, parmi eux, au moins neuf sont bien identifiés ; les autres allèles candidats attendent validation. L’analyse fonctionnelle des deux premières mutations rapportées, L764P et W887R [5], suggérait que la maladie résulte d’une haplo-insuffisance puisque ces mutants sont inactifs comme en témoigne leur incapacité à soutenir la croissance de cellules mammifères en culture. Depuis, nous avons clairement montré qu’au moins trois autres mutants, T345A, R689Q et M731T, sont fonctionnels et soutiennent la croissance cellulaire, mais affichent une cinétique altérée [6, 7].
Figure 2
Structure 2D schématique de la sous-unité catalytique Na,K-ATPase à 10 transmembranes montrant des mutations associées à la migraine hémiplégique [9].
![Structure 2D schématique de la sous-unité catalytique Na,K-ATPase à 10 transmembranes montrant des mutations associées à la migraine hémiplégique [9].](/fr/revues/ms/2006-v22-n4-ms1126/012797ar/media/012797arf002n.jpg)
Les mutations faux-sens fonctionnellement caractérisées sont surlignées (vert [5], jaune [6, 7]). Les mutations de délétion sont Del A (nt2897 et 2898 conduisant à une mutation de changement de phase et l’introduction d’un codon non-sens) et Del B (K935-S940 et insertion de I) et une mutation menant à une extension carboxy-terminale (Ext ; mutation codon non-sens X1021→R conduisant à l’ajout de 27 résidus à la partie carboxy-terminale).
Ainsi, l’ADNc des formes résistantes à l’ouabaïne des mutants[1] a été introduit dans des cellules HeLa cultivées en présence d’ouabaïne à 1µM afin d’inhiber l’activité de la pompe endogène humaine. De même que la forme de type sauvage, celles des mutants soutiennent la croissance des cellules HeLa suggérant que le phénotype de croissance, en présence de ouabaïne [5], n’est pas suffisant pour distinguer les allèles pathogéniques. Nous avons alors procédé à une analyse cinétique détaillée.
Nos résultats montrent que le mutant FHM2 T345A possède un taux de croissance et un renouvellement catalytique similaire à ceux de α2 de type sauvage. L'analyse d'autres paramètres cinétiques [6] suggère que la baisse d’affinité pour le K+ (doublement de K0.5(K)) reflète une augmentation du délestage de K+via les réactions E2.2[K]→E1.3Na (voir Figure 1). La modélisation par homologie [7] suggère que T345→A affecte la coordination du K+ en interférant avec le déplacement de TM4 lors des transitions de conformation contribuant à la formation de la poche de liaison des cations.
Inversement, R689Q et M731T entraînent plusieurs changements indicatifs d’un déplacement de l’équilibre E1↔E2 vers E1 [7] y compris une augmentation de l’affinité pour K+EXT, une baisse du renouvellement catalytique et une inhibition par l’orthovanadate inorganique, un analogue du phosphate inorganique se liant à E2. La modélisation structurale [7] prédit que R689→Q interfère avec les interactions normales des domaines cytoplasmiques, tandis que M731→T diminue l'affinité pour le Mg2+ dans la réaction de transfert qui dépend du groupement Mg2+-phosphoryl.
Ensemble, ces résultats suggèrent que le phénotype FHM est dû à une diminution de l’activité de la pompe α2 pouvant survenir soit par des mutations causant l’inactivation de la pompe (haplo-insuffisance [5]), une affinité apparente diminuée pour le K+ (T345A), soit par suite d’une baisse du renouvellement catalytique (R689Q et M731T). La diminution de l’activité entraînerait un délai dans la clairance de K+EXT et/ou un traitement/signalisation localisé altéré de Ca2+ secondaire à une activité réduite de l’échangeur Na+/Ca2+ colocalisé comme on le discute ci-dessous.
Puisque l’isoforme α2 de la Na,K-ATPase constitue un composant relativement mineur de l’activité totale de Na,K-ATPase dans le cerveau, l’explication la plus probable du phénotype de la maladie est que ces pompes sont confinées à des microdomaines de la membrane plasmique. Par conséquent, cette localisation conduirait à des changements dans les concentrations de cations dans des régions localisées à l’entrée des canaux ioniques et des échangeurs. Des études avec des souris déficientes en α2, montrant un accroissement local de la concentration d'ions Na+ et, via un échange de Na/Ca, une augmentation de la concentration d’ions Ca2+ près du réticulum endoplasmique [8], viennent étayer ce concept. La colocalisation de la sous-unité α2 de la pompe Na,K-ATPase et de l’échangeur Na/Ca de la membrane plasmique offre une base séduisante pour proposer que la FHM pourrait être causée par des mutations altérant le Ca2+ intracellulaire dans un microdomaine critique pour la signalisation de Ca2+ et ce, quel que soit le gène impliqué.
Parties annexes
Note
-
[1]
L’introduction des mutations Q116R et N127D à la bordure de la première boucle extracellulaire rend l’isoforme α2 et ses mutants relativement insensibles à l’ouabaïne afin de pouvoir distinguer leur cinétiques de ceux de l’enzyme endogène HeLa [10].
Références
- 1. Wessman M, Kaunisto M, Kallela M, Palotie A. The molecular genetics of migraine. Ann Med 2004 ; 36 : 462-73.
- 2. Dichgans M, Freilinger T, Eckstein G. et al. Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet 2005 ; 366 : 371-7.
- 3. Jorgensen PL, Kansson KOH, Karlish SJD. Structure and mechanism of Na, K-ATPase : functional sites and their interactions. Ann Rev Physiol 2003 ; 65 : 817-49.
- 4. Kaplan JH. Biochemistry of Na, K-ATPase. Ann Rev Biochem 2002 ; 71 : 511-35.
- 5. De Fusco M, Marconi R, Silvestri L, et al. Haploinsufficiency of ATP1A2 encoding the Na+/K+ pump alpha 2 subunit associated with familial hemiplegic migraine type 2. Nat Genet 2003 ; 33 : 192-6.
- 6. Segall L, Scanzano R, Kaunisto MA, et al. Kinetic alterations due to a missense mutation in the Na, K-ATPase alpha2 subunit cause familial hemiplegic migraine type 2. J Biol Chem 2004 ; 279 : 43692-6.
- 7. Segall L, Mezzetti A, Scanzano R, et al. Alterations in the alpha2 isoform of Na, K-ATPase associated with familial hemiplegic migraine type 2. Proc Natl Acad Sci USA 2005 ; 102 : 11106-11.
- 8. Golovina VA, Song H, James PF, et al. Na+ pump alpha 2-subunit expression modulates Ca2+ signaling. Am J Physiol 2003 ; 284 : C475-86.
- 9. Riant F, De Fusco M, Aridon P, et al. ATP1A2 mutations in 11 families with familial hemiplegic migraine. Hum Mutat 2005 ; 26 : 281.
- 10. Jewell EA, Lingrel JB. Comparison of the substrate dependence properties of the rat Na, K-ATPase alpha 1, alpha 2, and alpha 3 isoforms expressed in HeLa cells. J Biol Chem 1991 ; 266 : 16925-30.
Liste des figures
Figure 1
Le cycle de réaction de la Na,K-ATPase.
Selon le modèle « Albers-Post » abrégé, l’enzyme en E1 lie le Na+CYT et l’ATP avec haute affinité. L’enzyme est alors phosphorylé sur un résidu aspartate dans la boucle cytoplasmique TM4-TM5 (voir Figure 2) menant à l’occlusion des ions de Na+ (E1P.[3Na]). Par suite d’un changement de conformation en E2P, l’affinité apparente pour le K+EXT est nettement augmentée et diminuée pour le Na+ tel que ces derniers sont relâchés en faveur de deux K+EXT (E2P.2K). Les ions de K+ sont alors occlus en E2P.[2K] qui est rapidement déphosphorylée pour donner E2.[2K]. Les ions de K+ sont à leur tour relâchés, l’enzyme lie l’ATP (liaison à basse affinité) pour former E2.ATP qui change rapidement de conformation de sorte que l’enzyme est prêt pour un autre cycle. Le cycle catalytique de cette pompe ionique de type P implique une translocation de Na+ et K+ couplée à une hydrolyse ATP impliquant une phosphorylation et une déphosphorylation de la sous-unité catalytique α (résidu aspartyl sur l’enzyme, E) et des transitions conformationnelles de la phospho- et déphosphoenzyme (E1P→E2P et E1→E2). Ext : extracellulaire ; Cyt : cytoplasmique.
Figure 2
Structure 2D schématique de la sous-unité catalytique Na,K-ATPase à 10 transmembranes montrant des mutations associées à la migraine hémiplégique [9].
Les mutations faux-sens fonctionnellement caractérisées sont surlignées (vert [5], jaune [6, 7]). Les mutations de délétion sont Del A (nt2897 et 2898 conduisant à une mutation de changement de phase et l’introduction d’un codon non-sens) et Del B (K935-S940 et insertion de I) et une mutation menant à une extension carboxy-terminale (Ext ; mutation codon non-sens X1021→R conduisant à l’ajout de 27 résidus à la partie carboxy-terminale).