Résumés
Résumé
Le vieillissement est un processus qui continue à fasciner les biologistes de tous horizons, qu’ils s’intéressent à l’évolution, à la génétique, à la signalisation ou à la toxicité de l’environnement. De nombreuses théories, parfois contradictoires, sont proposées pour rendre compte des mécanismes du vieillissement, perçus par certains comme le résultat d’un programme inéluctable, par d’autres comme le fruit d’une suite d’agressions qui pourraient être évitées ou réparées. L’hypothèse « radicalaire » du vieillissement met au premier plan l’accumulation d’agressions oxydantes provoquées par les radicaux libres provenant principalement du métabolisme de l’oxygène et de l’azote. Cette hypothèse, proposée il y a une cinquantaine d’années, demeure l’une des plus populaires chez les spécialistes, même si certaines de ses prédictions n’ont pas été vérifiées de manière satisfaisante. Cet article présente les fondements de cette hypothèse, ses relations avec les autres théories, mitochondriales, métaboliques et génétiques, et la confronte à la réalité têtue des observations expérimentales pour proposer une vision plus intégrée des relations entre vieillissement et stress cellulaires.
Summary
A number of theories have attempted to account for ageing processes in various species. Following the « rate of living » theory of Pearl, Harman suggested fifty years ago that the accumulation of oxidants could explain the alteration of physical and cognitive functions with ageing. Oxygen metabolism leads to reactive species, including free radicals, which tend to oxydize surrounding molecules such as DNA, proteins and lipids. As a consequence various functions of cells and tissues can be altered, leading to DNA instability, protein denaturation and accumulation of lipid byproducts. Oxidative stress is an adaptive process which is triggered upon oxidant accumulation and which comprises the induction of protective and survival functions. Experimental evidence suggests that the ageing organism is in a state of oxidative stress, which supports the free radical theory. A number of other theories have been proposed ; some of these are actually compatible with the free radical theory. Caloric restriction is among the best models to increase life span in many species. While the relationship between caloric restriction and corrected metabolic rate is controversial, the decrease in ROS production by mitochondria appears to be experimentally supported. The ROS and mitochondrial theories of ageing appear to be compatible. Genetic models of increased life span, particularly those affecting the Foxo pathway, are usually accompanied by an increased resistance to oxidative insult. The free radical theory is not consistent with programmed senescence theories involving the cell division dependent decrease in telomere length ; however, oxidants are known to alter telomere structure. An appealing view of the role of oxidative stress in ageing is the trade-off principle which states that a phenotypic trait can be evolutionarily conserved because of its positive effects on development, growth or fertility, and despite its negative effect on somatic functions and ageing. It is likely that most cellular stresses which comprise adaptive and toxic functions follow such a rule.
Corps de l’article
La sagesse populaire qui recommande de se méfier de ses meilleurs amis s’applique parfaitement à l’oxygène, molécule dont on ne pourrait se passer au-delà de quelques minutes et qui est pourtant à l’origine de métabolites notoirement toxiques. Notre relation avec l’oxygène est cependant plus compliquée que cela et remonte aux origines de la vie. Il est bon de rappeler qu’il y a quelques milliards d’années, alors que l’atmosphère de la terre était très pauvre en oxygène, l’association de rayonnements solaires et d’eau provoquait déjà des espèces réactives oxygénées imposant sans doute aux organismes primitifs des mécanismes de détoxication. L’oxygène s’est, par la suite, accumulé grâce à la photosynthèse des cyanobactéries primitives, aux rayonnements et à divers évènements géologiques. Certains suggèrent, qu’en l’absence de systèmes de détoxication des espèces réactives oxygénées, il aurait été impossible à des organismes vivants de tirer, par la suite, tout le profit métabolique d’une atmosphère riche en oxygène. On pourrait donc parfaitement concevoir les formes de vie aérobie comme une adaptation à un déchet toxique, l’oxygène et à ses métabolites. Les réactions chimiques et les enzymes qui constituent ce métabolisme sont présentes pratiquement dès les origines de la vie [1].
Origine et destinée des ERO (espèces réactives de l’oxygène)
La molécule de dioxygène est en réalité bi-radicalaire. Elle possède, en effet, deux électrons célibataires sur des orbitales différentes. Le dioxygène est susceptible de récupérer quatre électrons, mais ses capacités oxydantes sont limitées par une barrière cinétique importante. En présence de rayonnements, de métaux ou d’enzymes, il est capable de capter un électron pour donner le radical superoxyde O·̱2 qui est un radical modérément réactif (Figure 1). Ce radical est le substrat d’enzymes essentielles, les superoxyde dismutases (SOD), qui le transforment en eau oxygénée H2O2. L’eau oxygénée peut avoir plusieurs destinées. En présence de métaux, en particulier de fer Fe++, elle est transformée en radical hydroxyl .OH par la réaction de Fenton. Ce dernier est extrêmement réactif et va oxyder très rapidement les molécules voisines, formant parfois d’autres radicaux libres [2, 3]. L’eau oxygénée peut aussi subir des réactions de détoxication catalysées par la catalase, la glutathion peroxydase ou les peroxyrédoxines. De même, plusieurs composés, notamment les vitamines E et C, peuvent interagir avec les radicaux et éviter leur accumulation [4] (Figure 1).
Figure 1
Voie métabolique de l’oxygène et des ERO (espèces réactives de l’oxygène).
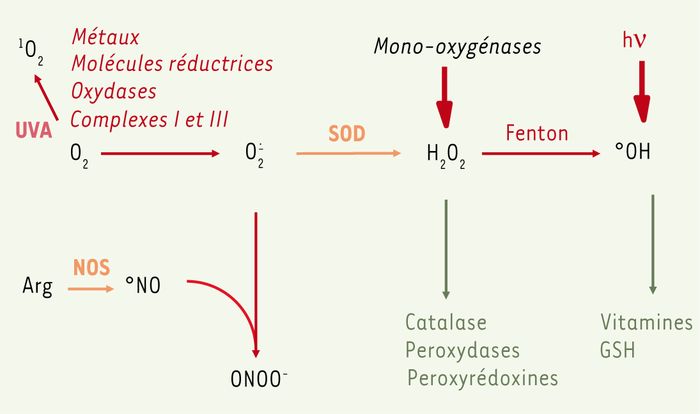
L’oxygène peut subir des étapes successives de réduction conduisant à la formation d’ERO. En présence de métaux, de molécules organiques ou d’enzymes (oxydases ou complexes de la chaîne respiratoire), il est capable de capter un électron pour donner le radical superoxyde O2.-. Ce radical est le substrat d’enzymes essentielles, les superoxyde dismutases (SOD) qui le transforment en eau oxygénée H2O2. L’eau oxygénée peut avoir plusieurs destinées. En présence de métaux, en particulier le fer Fe2+, elle est transformée en radical hydroxyl °OH par la réaction de Fenton. Ce dernier est extrêmement réactif et va oxyder très rapidement les molécules voisines formant parfois d’autres radicaux libres. L’eau oxygénée peut aussi subir des réactions de détoxication catalysées par la catalase, la glutathion peroxydase ou les peroxyrédoxines. De même, plusieurs composés, notamment les vitamines E et C et le glutathion, peuvent interagir avec les radicaux et les détoxiquer. Sous l’influence de rayonnements UV, l’oxygène peut être transformé en oxygène singulet. Le métabolisme de l’oxygène croise celui de l’azote puisque O2.- interagit avec un autre radical, le monoxyde d’azote NO et conduit au composé toxique, le peroxynitrite ONOO-. Arg : arginine ; NOS : NO synthase.
Les ERO peuvent être produites par des agents physiques comme les rayonnements, des réactions chimiques et surtout enzymatiques. En effet, toute réaction impliquant de l’O2 et un système réducteur de transfert d’électrons est susceptible de libérer des ERO. C’est ainsi que la chaîne respiratoire provoque une libération importante d’ERO, mais dont l’intensité demeure controversée (voir plus loin). D’autres activités enzymatiques fournissent aussi des ERO, notamment les NADPH oxydases au cours de l’inflammation et les cytochromes P450 au cours de la détoxication des xénobiotiques. Ainsi, la mitochondrie, la membrane plasmique et le réticulum endoplasmique sont les sièges principaux de libération d’ERO [5] (Figure 2).
Figure 2
Origines et actions des ERO.
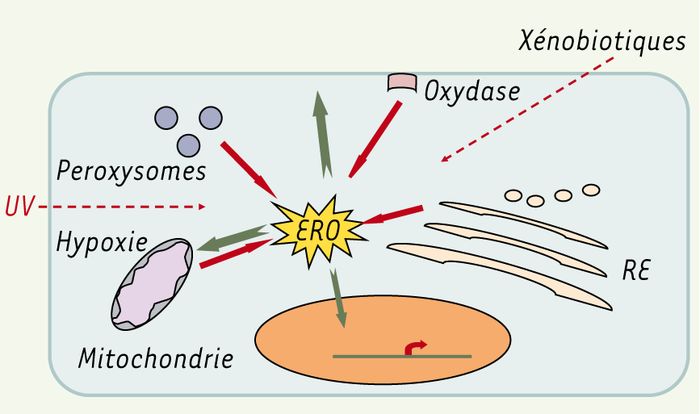
Les ERO peuvent provenir de différents compartiments cellulaires : la mitochondrie, même en cas d’hypoxie (chaîne respiratoire), le réticulum endoplasmique (RE) (mono-oxygénases), la membrane plasmique (oxydases), les peroxysomes et le cytoplasme. Ils peuvent être produits directement par des rayonnements, des molécules endogènes ou des xénobiotiques. Ils exercent des effets sur les acides nucléiques, ARN, ADN nucléaire et mitochondrial (génotoxicité), sur les lipides membranaires, conduisant à des intermédiaires toxiques, sur les protéines à des niveaux différents allant jusqu’à la carbonylation et la dénaturation, et sur d’autres composants cellulaires.
Il existe, dans la cellule, d’autres oxydants très puissants, qu’ils soient des radicaux libres ou non [6]. Par exemple, des oxydants chlorés (HOCl) sont libérés par les macrophages et ont une activité bactéricide importante. Par ailleurs, le monoxyde d’azote (NO) est un radical libre qui est surtout réputé pour ses propriétés physiologiques. Or, le NO interagit avec l’anion superoxyde pour donner le peroxynitrite, composé extrêmement réactif et toxique. NO et peroxynitrite interagissent avec des protéines et peuvent altérer leurs propriétés. D’autres molécules comme les hydroquinones se retrouvent sous forme de radicaux libres après leur réaction avec le radical °OH, et, de par leur structure, stabilisent leur électron célibataire (radical semi-quinonique). Elles sont ainsi susceptibles de diffuser dans la cellule et d’oxyder d’autres molécules à distance, propageant ainsi une chaîne de réactions radicalaires.
La double vie des ERO
Il serait erroné de ne voir les ERO que sous l’angle de leur toxicité. Les ERO, et plus généralement les réactions d’oxydo-réduction, jouent un rôle physiologique considérable, en particulier dans les cascades de signalisation [7]. Le NO en est un exemple classique, puisqu’en activant la guanylate cyclase cytosolique, il exerce des fonctions physiologiques dans le système vasculaire, immunitaire, neuronal et métabolique. Il en est de même de l’anion superoxyde et de l’eau oxygénée qui activent plusieurs voies de signalisation comme la voie NFκB, Nrf-2, P53, JNK et P38 MAPK. Ces composés jouent un rôle crucial au cours de l’inflammation et de l’équilibre entre la croissance, l’apoptose et la sénescente cellulaires. La production des ERO est stimulée par des hormones, des facteurs de croissance et des cytokines [8].
À ces fonctions biologiques, la réactivité particulière des ERO ajoute des propriétés toxiques importantes et diversifiées. En effet, toutes les macromolécules cellulaires sont des cibles potentielles des ERO. Ainsi, les bases nucléiques sont susceptibles d’être oxydées, conduisant notamment à la formation de 8-oxo-guanine, à l’origine de mutations géniques. C’est sans doute ce qui explique la génotoxicité des radicaux libres [8]. Notons que l’ADN mitochondrial, du fait de sa proximité avec la chaîne respiratoire, site éminent de production des ERO, et du fait de l’absence de chromatine organisée, est particulièrement exposé à l’oxydation [9]. Les protéines sont aussi la cibles des ERO, en particulier certains acides aminés comme la cystéine, la méthionine et la tyrosine. Cette oxydation joue un rôle dans la maturation et dans la signalisation mais peut aussi conduire à une toxicité cellulaire. Une oxydation plus franche des protéines peut conduire à leur carbonylation et à leur dénaturation. Les ERO réagissent aussi avec les acides gras insaturés, conduisant à la formation d’hydroperoxydes. Ils contribuent aussi à la glycation des protéines et à la formation des dérivés de cette glycation qu’on appelle les AGE (advanced glycation endproducts). De par leur réactivité et leurs cibles, les ERO ont été incriminés dans la pathogénie d’un grand nombre de pathologies comme le cancer, les maladies cardiovasculaires, les maladies dégénératives et le diabète. Notons que ces pathologies sont intimement associées au vieillissement.
Un des obstacles pour bien distinguer la part physiologique et la part toxique des effets des ERO est la difficulté de les mesurer de manière fiable. Des techniques de RPE (résonance paramagnétique électronique) et de fluorescence sont disponibles mais elles sont parfois difficiles à mettre en oeuvre. Par ailleurs, la demi-vie des ERO est très courte et leur production peut être compartimentée. On en est ainsi réduit à doser des marqueurs biologiques d’oxydation par les ERO, en particulier des marqueurs lipidiques, des dialdéhydes, la 8-oxo-guanine, les AGE ou la carbonylation des protéines. Ce sont des marqueurs indirects de l’action des ERO.
Qu’est-ce qu’un stress oxydant ?
Le stress oxydant est communément défini comme un déséquilibre entre les systèmes oxydants et les capacités anti-oxydantes d’un organisme, d’une cellule ou d’un compartiment cellulaire. Cette définition ne rend pas justice à la notion de stress qui est avant tout une réponse à une modification des conditions habituelles de vie cellulaire. Lorsque des ERO commencent à s’accumuler dans la cellule, ils peuvent être neutralisés par des molécules de défense anti-oxydantes présentes dans la cellule comme le glutathion, les vitamine E et C, la bilirubine, l’acide lipoïque, et des enzymes comme la catalase, la superoxyde dismutase, la glutathion peroxydase, les peroxyrédoxines… [8-10]. Dans un premier temps, la cellule ne modifie pas ses propriétés biologiques. Si les ERO continuent à s’accumuler, une adaptation plus consistante de la cellule est nécessaire avec l’induction de gènes codant des enzymes anti-oxydantes, des protéines chaperons, des enzymes impliquées dans la réparation de l’ADN et des protéines. On observe aussi une répression des systèmes susceptibles de libérer des ERO, notamment la chaîne respiratoire, les cytochromes P450 et la NADPH oxydase [3, 5]. Dans ces conditions, nous pouvons parler de stress dans la mesure où la cellule a adapté ses fonctions biologiques, notamment son expression génique, aux modifications de son environnement (Figure 3). Souvent, l’induction des enzymes anti-oxydantes est perçue comme le révélateur de l’existence d’un stress oxydant, même si ceci a été remis partiellement en cause dans des expériences de génomique récentes [11]. À un stade ultime, la cellule peut suivre la voie de l’apoptose ou de la sénescence.
Figure 3
La double vie des ERO : pléiotropie contradictoire et compromis.

Les ERO ont des origines multiples et exercent deux types d’action : des effets physiologiques au cours de la croissance ou de la défense de l’organisme et des effets délétères affectant différentes macromolécules et pouvant s’accumuler avec l’âge pour conduire à des pathologies et aux manifestations du vieillissement. Ces actions contradictoires sont tolérées grâce au stress oxydant qui est la réponse cellulaire adaptative à l’excès d’ERO. Au cours de l’évolution, les fonctions physiologiques des ERO auraient permis leur maintien malgré l’accumulation de leur toxicité avec l’âge.
Selon cette définition, il est aisé de détecter un stress oxydant provoqué par une élévation aiguë des ERO. La situation est plus complexe lorsque l’on recherche un stress oxydant chronique au cours duquel, d’une part, les élévations des ERO sont atténuées par des boucles de régulation, et, d’autre part, les inductions des enzymes anti-oxydantes et réparatrices sont plus modestes, ces enzymes étant parfois elles-mêmes altérées par oxydation.
Bref aperçu de la théorie radicalaire du vieillissement
Le vieillissement s’accompagne d’une altération globale d’un ensemble de fonctions physiologiques ainsi que d’une susceptibilité plus élevée face à différentes maladies. La théorie radicalaire explique ces altérations par l’accumulation de molécules oxydées et par les conséquences de cette oxydation comme l’apparition de mutations, la carbonylation des protéines, leur dénaturation et leur agrégation, l’oxydation des lipides et l’augmentation des AGE. Cette théorie a été formulée pour la première fois par Harman, en 1956 [12], avant même que la pertinence biologique des radicaux libres eût été démontrée. La mise en évidence, par Fridovitch, en 1969, de la superoxyde dismutase dont le substrat unique est le radical superoxyde, a contribué à établir la réalité biologique des radicaux libres et leurs rôles possibles physiologiques ou physiopathologiques [13]. Plusieurs arguments militent en faveur de l’implication des radicaux libres dans les mécanismes du vieillissement. Une élévation des marqueurs biologiques du stress oxydant comme la 8-oxo-guanine, le dialdéhyde malonique (MDA) et les isoprostanes a été observée au cours du vieillissement de nombreuses espèces [1, 10]. Par ailleurs, des études récentes de la variation du transcriptome au cours du vieillissement, y compris chez le singe, ont révélé l’induction de plusieurs gènes codant des enzymes anti-oxydantes et la répression de gènes de la chaîne respiratoire, révélatrices d’une adaptation au long cours à un état cellulaire pro-oxydant [14]. Enfin, la détérioration des macromolécules cellulaires est compatible avec une élévation de la pression oxydante avec l’âge puisque l’on note une augmentation des mutations et des délétions, en particulier de l’ADN mitochondrial, une carbonylation des protéines et leur dénaturation, une élévation des produits de glycation avancée (AGE) révélatrice d’un état prédiabétique ou diabétique [10, 15, 16]. Enfin, l’efficacité des mécanismes de réparation cellulaire comme le protéasome, les protéines chaperons, plusieurs enzymes réductrices et les sytèmes de réparation de l’ADN diminuent avec l’âge, ce qui contribue à la fixation et à l’accumulation des anomalies [17, 18].
Restriction calorique, mitochondrie et stress oxydant
La longévité de très nombreuses espèces comme le ver C. elegans ou la souris est prolongée par la restriction calorique [19, 20]. Chez l’homme, des travaux conduits sur la population japonaise de l’île d’Okinawa qui comprend un nombre particulièrement élevé de centenaires, indique aussi que la restriction calorique - qu’il ne faut pas confondre avec des déficits nutritionnels - pourrait aussi prolonger la durée de vie chez l’homme [1]. L’hypothèse la plus répandue est que la restriction calorique diminue le métabolisme basal, et donc la consommation d’oxygène. Cette explication fait écho aux idées défendues par Pearl dès les années 20, suggérant que l’activité métabolique globale est inversement corrélée à la durée de vie des espèces et des individus [21]. Cette hypothèse ne semble pas vérifiée chez toutes les espèces, en particulier les oiseaux qui possèdent un métabolisme très actif et une durée de vie supérieure à celle d’autres espèces [22]. Le modèle de la restriction calorique a été considéré comme parfaitement compatible avec la théorie radicalaire du vieillissement, dans la mesure où la consommation d’oxygène par la mitochondrie s’accompagne d’un relargage d’ERO par la chaîne respiratoire. Le taux d’ERO relargué est très controversé puisque nous sommes passés d’estimation (réalisées in vitro) d’ERO produits allant de 2 à 4 % de l’oxygène consommé à des estimations plus réalistes de 0,2 à 0,4 % [15]. Néanmoins, on peut comprendre qu’une diminution de la consommation d’oxygène s’accompagne d’une diminution de la production mitochondriale d’ERO et donc, selon la théorie radicalaire du vieillissement, d’un prolongement de la durée de vie. Il n’en demeure pas moins qu’une activité métabolique forte chez certaines espèces ne s’accompagne pas d’une diminution de la longévité [22, 23]. Pour comprendre ce paradoxe, il faut remettre en cause la relation entre consommation d’oxygène et production d’ERO. Dans certaines conditions, comme l’exercice physique, une activité métabolique importante ne s’accompagne pas d’une élévation proportionnelle de la production d’ERO. Parmi les mécanismes qui peuvent modifier la relation entre la production d’ERO et la consommation d’oxygène, le découplage mitochondrial réalisé par les protéines découplantes UCP semble être au premier plan. Les UCP, dont certaines isoformes sont induites par le stress oxydant, réduisent le gradient de protons au niveau de la membrane mitochondriale interne, et, de ce fait, diminueraient la production d’ERO [23, 24]. Ce rôle des UCP demeure cependant controversé.
Il est sans doute un peu réducteur de ne voir dans les effets de la restriction calorique que ceux qui sont liés à la résistance aux agressions oxydantes. Des effets hormonaux et métaboliques globaux, en particulier l’amélioration de la sensibilité à l’insuline, sont sans doute essentiels [23]. Mais nous verrons ultérieurement que cette piste pourrait aussi rejoindre celle des radicaux libres.
Modèles génétiques
Les expériences de transgenèse et de délétion génique visant à altérer la longévité de différentes espèces sont dans l’ensemble cohérentes avec un rôle primordial des ERO dans les mécanismes de vieillissement [25, 26]. Une des voies de signalisation la plus clairement corrélée avec la longévité est celle de l’insuline/IGF. Les premières expériences chez le ver C. elegans ont montré que plusieurs acteurs de cette voie de signalisation sont impliqués dans la longévité. Cette voie aboutit au facteur Daf 16, équivalent des facteurs transcriptionnels de mammifère Foxo. La plupart des mutants ayant une durée de vie augmentée sont plus résistants à une agression oxydante. Cette observation est cohérente avec les effets de Foxo qui contrôle l’expression de nombreux gènes anti-oxydants (Figure 4). Notons cependant que certains mutants de C. elegans ne semblent pas modifier la résistance à l’oxydation et qu’il est difficile, à ce stade, de corréler de manière complète l’extension de la longévité et la résistance aux dommages oxydants. Le rôle de la voie des IGF a été confirmé chez la souris par l’équipe de Martin Holzenberger, qui, travaillant sur les récepteurs de l’IGF-1, a démontré que les hétérozygotes IGF-1R+/- ont une durée de vie plus longue que les sauvages et résistent mieux à une agression oxydante [27].
Figure 4
Théorie radicalaire et autres théories du vieillissement.
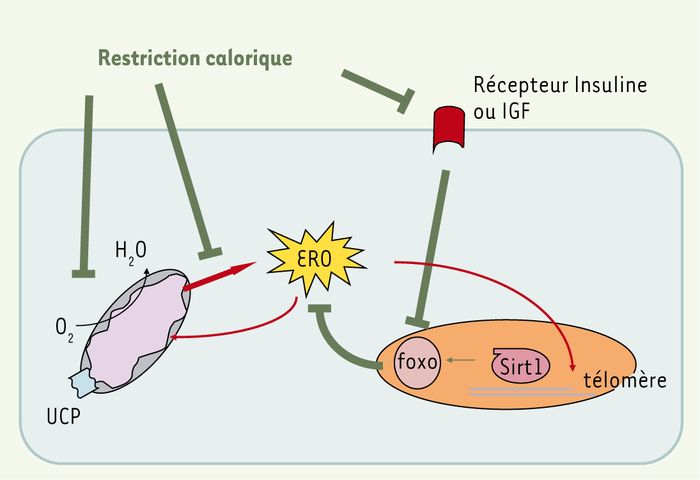
L’implication des ERO dans le vieillissement n’est pas nécessairement contradictoire avec d’autres théories. Ainsi, les ERO peuvent altérer les télomères, contribuant ainsi à leur raccourcissement et à la sénescence cellulaire. Différents modèles génétiques impliquent la voie de signalisation insuline/IGF qui aboutit au facteur transcriptionnel Foxo. Or, ce facteur induit des gènes impliqués dans la résistance aux ERO et dans le stress oxydant. Par ailleurs, chez les mammifères, ce facteur est lui-même activé par les protéines Sirt, elles-mêmes impliquées dans la stabilité télomérique et la sénescence cellulaire. La restriction calorique est sans doute le modèle non génétique d’accroissement de la longévité le plus connu. La restriction calorique peut d’une part affecter la voie insuline/IGF/Foxo, et, d’autre part, le métabolisme basal et les fonctions mitochondriales, autres acteurs essentiels des théories du vieillissement. Les effets de la restriction calorique sur la consommation d’oxygène corrigée sont discutés. Néanmoins, la production d’ERO par la mitochondrie semble bien diminuer ; les protéines UCP interviendraient dans cet effet. Ainsi, la théorie radicalaire s’avère complémentaire et compatible avec d’autres visions du vieillissement.
Le gène sir2 a suscité depuis quelques années un intérêt croissant : il a été découvert dans la levure puis dans de nombreuses autres espèces. Il joue un rôle dans le silencing ou la répression génique et code pour une histone désacétylase qui possède la remarquable propriété d’être dépendante du NAD [28]. Plusieurs auteurs ont montré que Sir2 ou ses équivalents sont requis pour l’accroissement de la longévité de la drosophile et de C. elegans par la restriction calorique, ce qui a permis d’établir un lien supplémentaire entre la longévité et le métabolisme énergétique qui a un impact direct sur le rapport NAD/NADH [29]. La relation entre la voie des Sir2 et le stress oxydant n’est pas évidente. Cependant, les orthologues de Sir2 chez les mammifères, notamment Sirt1, ont une activité désacétylase moins spécifique qui affecte aussi des facteurs nucléaires comme P53 et surtout Foxo. Contrairement à Sir2 chez la levure, l’expression de Sirt1 chez la souris contribue à augmenter les défenses anti-oxydantes dans cette espèce [30].
Anti-oxydants : le point faible et l’espoir
Les détracteurs de la théorie des radicaux libres relèvent que l’une des prédictions principales de cette théorie, à savoir un effet bénéfique des anti-oxydants sur le vieillissement, n’est pas vérifiée, sans que cela empêche une exploitation commerciale acharnée de ces composés. Il est vrai que l’échec de certains essais cliniques a soulevé des doutes sur l’importance du stress oxydant dans le vieillissement et la survenue de pathologies [10]. Il faut cependant prendre en considération les points suivants pour mieux comprendre ce débat. Le stress oxydant a des effets pléiotropes et il est difficile de déterminer l’effet toxique le plus important - et le moment auquel il est le plus critique - pour suivre son évolution en fonction du traitement. Par ailleurs, la notion d’anti-oxydant a ses limites, puisqu’une molécule peut se comporter comme un oxydant ou un anti-oxydant selon son partenaire moléculaire et selon les conditions cellulaires [31]. Enfin, il est sans doute excessif de considérer le stress oxydant comme la cause unique du vieillissement ou de la survenue de pathologies associées à l’âge, et en conséquence, un traitement anti-oxydant devrait être considéré principalement comme un adjuvant.
Malgré ces restrictions, des travaux expérimentaux récents soutiennent le rôle du stress oxydant au cours du vieillissement. Ainsi, la surexpression simultanée de la superoxyde dismutase et de la catalase retarde le vieillissement chez la drosophile [32]. Nous avons vu ci-dessus que l’association de ces deux enzymes était nécessaire pour la détoxication de l’anion superoxyde. Un travail récent indique que la longévité de la souris pouvait être allongée grâce à la surexpression de la catalase spécifiquement dans la mitochondrie [33]. Enfin, des travaux du laboratoire de Bruce Ames suggèrent que l’association entre l’acide lipoïque qui augmente les défenses anti-oxydantes et l’acétyl-carnitine, qui améliore les fonctions énergétiques de la mitochondrie âgée, a des effets bénéfiques sur certains marqueurs moléculaires et métaboliques du vieillissement [34]. L’intérêt de ces quelques exemples est de montrer qu’une bonne connaissance des mécanismes anti-oxydants et de l’interaction entre métabolisme et stress oxydant, peut aider à concevoir des moyens thérapeutiques plus efficaces et à tester de manière plus pertinente la relation entre vieillissement et stress oxydant.
Le vieillissement est-il programmé ?
Il est important de confronter la théorie radicalaire aux autres théories du vieillissement exposées ailleurs. Certains spécialistes de l’évolution ont émis l’hypothèse d’un avantage sélectif du vieillissement qui permettrait, chez certaines espèces, dans des conditions de restriction nutritive, de favoriser les individus les plus jeunes et les plus susceptibles de se reproduire par rapport aux plus âgés. Kirkwood et d’autres auteurs ont critiqué la notion d’avantage sélectif du vieillissement profitant à l’espèce aux dépens de l’individu [18]. Nous ne discuterons pas ici de cette controverse qui sort du cadre de cet article. Notons cependant que les théories de vieillissement programmé ne sont pas nécessairement contradictoires avec la théorie oxydative. Par exemple, le programme de sénescence cellulaire reposant sur une diminution progressive de la taille des télomères à chaque division cellulaire peut être amplifié par l’altération de ces télomères provoquée par le stress oxydant (Figure 4). Il semble improbable que le vieillissement soit uniquement dicté par un programme sélectionné au cours de l’évolution, et, dans tous les cas, il pourrait agir de manière synergique avec le stress oxydant.
ERO entre pléiotropie et compromis
L’implication du stress oxydant dans le vieillissement s’intègre bien dans un concept plus général qu’on peut appeler « pléiotropie antagoniste » (antagonistic pleiotropy) qui considère que les altérations associées au vieillissement sont la conséquences de l’activité de gènes essentiels pour le développement et la fertilité des organismes [18]. L’exemple le plus cité est celui de la protéine p53 dont les fonctions protectrices sont reconnues, mais qui est impliquée dans les phénomènes de sénescence. De nombreux autres facteurs peuvent sans doute relever de cette catégorie. Il y aurait donc un compromis permettant de sélectionner ces gènes pour leur effets bénéfiques au début de la vie tout en tolérant leur toxicité possible apparaissant avec l’âge. On peut proposer que la toxicité des oxydants et des radicaux libres qui s’accumulent avec l’âge est le prix à payer pour leurs effets bénéfiques, par exemple dans la signalisation et les défenses de l’organisme, ou tout simplement parce qu’ils sont générés de manière difficile à éviter au cours de processus essentiels comme la respiration et le métabolisme (Figure 3).
Le capital stress
Il est sans doute utile de replacer le stress oxydant dans le cadre plus général des stress cellulaires qui sont tous des mécanismes d’adaptation à des agressions qui peuvent engendrer des conséquences toxiques [35]. Les enzymes du métabolisme des xénobiotiques, qui jouent un rôle de premier plan dans l’adaptation à un stress chimique, peuvent elles-mêmes produire des intermédiaires métaboliques très toxiques. Il y a donc là aussi un compromis entre une fonction nécessaire et des risques toxiques. Ce risque demeure limité tant que les mécanismes de réparation restent fonctionnels. La réparation des altérations de l’ADN en sont un bel exemple, mais on pourrait dire la même chose de la réparation des protéines, et, à l’échelle de l’organisme, de l’élimination des cellules altérées par apoptose et même de l’inflammation [18, 36]. Lorsque les systèmes de réparation eux-mêmes sont atteints, il est probable qu’un cap est passé en direction de la détérioration des fonctions cellulaires. Tout se passe comme si on avait un « capital stress » qui, lorsqu’il est épuisé, conduit au vieillissement. Un des arguments en faveur de cette hypothèse est que les mutations dans les enzymes de réparation de l’ADN s’accompagnent d’un vieillissement accéléré [37]. Cela est également compatible avec les observations montrant que l’accumulation de protéines agrégées dans une cellule épuise les systèmes de réparation conformationnelle et le protéasome. Ainsi, toute condition environnementale ou génétique qui augmente l’exposition à des agressions (oxydantes ou non) ou qui diminue les capacités de défenses ou de réparation, devrait se traduire par un vieillissement accéléré. Les nombreux modèles génétiques disponibles ou en cours de réalisation devraient permettre de tester cette hypothèse.
Conclusions
La théorie des radicaux libres est soutenue par des arguments expérimentaux dont la plupart sont corrélatifs même si des expériences récentes présentent des preuves plus directes. Elle me semble compatible avec les autres théories dans la mesure où elle ne vise pas à expliquer l’ensemble des phénomènes liés au vieillissement ; en effet, certains tissus peuvent souffrir de la limitation du nombre de divisions cellulaires alors que d’autres seront plus sensibles à l’atteinte de cellules non réplicatives. Par ailleurs, le stress oxydant gagnerait à être remis dans le contexte des stress cellulaires dans leur ensemble qui, eux aussi, conduisent à l’accumulation d’altérations. Il est intéressant de noter d’ailleurs que le stress oxydant accompagne pratiquement l’ensemble des stress cellulaires.
Parties annexes
Références
- 1. Lane N. Oxygen, the molecule that made the world. New York : Oxford University Press, 2003 : 366 p.
- 2. Gardès-Albert M, Jore D. Aspects physicochimiques des radicaux libres centrés sur l’oxygène. In : Delattre JB, Bonnefont-Rousselot D, eds. Radicaux libres et stress oxydant. Aspects biologiques et pathologiques. Paris : Lavoisier, 2005 : 1-23.
- 3. Morel Y, Barouki R. Repression of gene expression by oxidative stress. Biochem J 1999 ; 342 : 481-96.
- 4. Roussel AM, Nève J, Hininger I. Antioxydants et nutrition. In : Delattre JB, Bonnefont-Rousselot D, eds. Radicaux libres et stress oxydant. Aspects biologiques et pathologiques. Paris : Lavoisier, 2005 : 261-80.
- 5. Barouki R, Morel Y. Repression of cytochrome P450 1A1 gene expression by oxidative stress : mechanisms and biological implications. Biochem Pharmacol 2001 ; 61 : 511-6.
- 6. Beaudeux JL, Vasson MP. Sources cellulaires des espèces réactives de l’oxygène. In : Delattre JB, Bonnefont-Rousselot D, eds. Radicaux libres et stress oxydant. Aspects biologiques et pathologiques. Paris : Lavoisier, 2005 : 45-86.
- 7. Finkel T. Oxidant signals and oxidative stress. Curr Opin Cell Biol 2003 ; 15 : 247-54.
- 8. Morel Y, Barouki R. Influence du stress oxydant sur la régulation des gènes. Med Sci(Paris) 1998 ; 14 : 713-21.
- 9. Beckman KB, Ames BN. Endogenous oxidative damage of mtDNA. Mutat Res 1999 ; 424 : 51-8.
- 10. Delattre J, Thérond P, Bonnefont-Rousselot D. Espèces réactives de l’oxygène, antioxydants et vieillissement. In : Delattre JB, Bonnefont-Rousselot D, eds. Radicaux libres et stress oxydant. Aspects biologiques et pathologiques. Paris : Lavoisier, 2005 : 281-309.
- 11. Desaint S, Luriau S, Aude JC, et al. Mammalian antioxidant defenses are not inducible by H2O2. J Biol Chem 2004 ; 279 : 31157-63.
- 12. Harman D. Aging : a theory based on free radical and radiation chemistry. J Gerontol 1956 ; 11 : 298-300.
- 13. McCord JM, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J Biol Chem 1969 ; 244 : 6049-55.
- 14. Kayo T, Allison DB, Weindruch R, Prolla TA. Influences of aging and caloric restriction on the transcriptional profile of skeletal muscle from rhesus monkeys. Proc Natl Acad Sci USA 2001 ; 98 : 5093-8.
- 15. Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell 2005 ; 120 : 483-95.
- 16. Berlett BS, Stadtman ER. Protein oxidation in aging, disease, and oxidative stress. J Biol Chem 1997 ; 272 : 20313-6.
- 17. Sohal RS, Mockett RJ, Orr WC. Mechanisms of aging : an appraisal of the oxidative stress hypothesis. Free Radic Biol Med 2002 ; 33 : 575-86.
- 18. Kirkwood TB. Understanding the odd science of aging. Cell 2005 ; 120 : 437-47.
- 19. Sohal RS, Weindruch R. Oxidative stress, caloric restriction, and aging. Science 1996 ; 273 : 59-63.
- 20. Heilbronn LK, Ravussin E. Calorie restriction and aging : review of the literature and implications for studies in humans. Am J Clin Nutr 2003 ; 78 : 361-9.
- 21. Pearl R. The rate of living. New York : Knopf, 1928.
- 22. Barja G. Mitochondrial free radical production and aging in mammals and birds. Ann NY Acad Sci 1998 ; 854 : 224-38.
- 23. Bordone L, Guarente L. Calorie restriction, SIRT1 and metabolism : understanding longevity. Nat Rev Mol Cell Biol 2005 ; 6 : 298-305.
- 24. Pecqueur C, Alves-Guerra MC, Gelly C, et al. Uncoupling protein 2, in vivo distribution, induction upon oxidative stress, and evidence for translational regulation. J Biol Chem 2001 ; 276 : 8705-12.
- 25. Guarente L, Kenyon C. Genetic pathways that regulate ageing in model organisms. Nature 2000 ; 408 : 255-62.
- 26. Purdom S, Chen QM. Linking oxidative stress and genetics of aging with p66Shc signaling and forkhead transcription factors. Biogerontology 2003 ; 4 : 181-91.
- 27. Holzenberger M, Dupont J, Ducos B, et al. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 2003 ; 421 : 182-7.
- 28. Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 2000 ; 403 : 795-800.
- 29. Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev 1999 ; 13 : 2570-80.
- 30. Brunet A, Sweeney LB, Sturgill JF, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004 ; 303 : 2011-5.
- 31. Gutteridge JM, Halliwell B. Free radicals and antioxidants in the year 2000. A historical look to the future. Ann NY Acad Sci 2000 ; 899 : 136-47.
- 32. Orr WC, Sohal RS. Extension of life-span by overexpression of superoxide dismutase and catalase in Drosophila melanogaster. Science 1994 ; 263 : 1128-30.
- 33. Schriner SE, Linford NJ, Martin GM, et al. Extension of murine lifespan by overexpression of catalase targeted to mitochondria. Science 2005 ; 308 : 1909-11.
- 34. Hagen TM, Liu J, Lykkesfeldt J, et al. Feeding acetyl-L-carnitine and lipoic acid to old rats significantly improves metabolic function while decreasing oxidative stress. Proc Natl Acad Sci USA 2002 ; 99 : 1870-5.
- 35. Barouki R. La cellule stressée. Med Sci(Paris) 1999 ; 15 : 1359-61.
- 36. Petropoulos I, Friguet B. Protein maintenance in aging and replicative senescence : a role for the peptide methionine sulfoxide reductases. Biochim Biophys Acta 2005 ; 1703 : 261-6.
- 37. Lombard DB, Chua KF, Mostoslavsky R, et al. DNA repair, genome stability, and aging. Cell 2005 ; 120 : 497-512.
Liste des figures
Figure 1
Voie métabolique de l’oxygène et des ERO (espèces réactives de l’oxygène).
L’oxygène peut subir des étapes successives de réduction conduisant à la formation d’ERO. En présence de métaux, de molécules organiques ou d’enzymes (oxydases ou complexes de la chaîne respiratoire), il est capable de capter un électron pour donner le radical superoxyde O2.-. Ce radical est le substrat d’enzymes essentielles, les superoxyde dismutases (SOD) qui le transforment en eau oxygénée H2O2. L’eau oxygénée peut avoir plusieurs destinées. En présence de métaux, en particulier le fer Fe2+, elle est transformée en radical hydroxyl °OH par la réaction de Fenton. Ce dernier est extrêmement réactif et va oxyder très rapidement les molécules voisines formant parfois d’autres radicaux libres. L’eau oxygénée peut aussi subir des réactions de détoxication catalysées par la catalase, la glutathion peroxydase ou les peroxyrédoxines. De même, plusieurs composés, notamment les vitamines E et C et le glutathion, peuvent interagir avec les radicaux et les détoxiquer. Sous l’influence de rayonnements UV, l’oxygène peut être transformé en oxygène singulet. Le métabolisme de l’oxygène croise celui de l’azote puisque O2.- interagit avec un autre radical, le monoxyde d’azote NO et conduit au composé toxique, le peroxynitrite ONOO-. Arg : arginine ; NOS : NO synthase.
Figure 2
Origines et actions des ERO.
Les ERO peuvent provenir de différents compartiments cellulaires : la mitochondrie, même en cas d’hypoxie (chaîne respiratoire), le réticulum endoplasmique (RE) (mono-oxygénases), la membrane plasmique (oxydases), les peroxysomes et le cytoplasme. Ils peuvent être produits directement par des rayonnements, des molécules endogènes ou des xénobiotiques. Ils exercent des effets sur les acides nucléiques, ARN, ADN nucléaire et mitochondrial (génotoxicité), sur les lipides membranaires, conduisant à des intermédiaires toxiques, sur les protéines à des niveaux différents allant jusqu’à la carbonylation et la dénaturation, et sur d’autres composants cellulaires.
Figure 3
La double vie des ERO : pléiotropie contradictoire et compromis.
Les ERO ont des origines multiples et exercent deux types d’action : des effets physiologiques au cours de la croissance ou de la défense de l’organisme et des effets délétères affectant différentes macromolécules et pouvant s’accumuler avec l’âge pour conduire à des pathologies et aux manifestations du vieillissement. Ces actions contradictoires sont tolérées grâce au stress oxydant qui est la réponse cellulaire adaptative à l’excès d’ERO. Au cours de l’évolution, les fonctions physiologiques des ERO auraient permis leur maintien malgré l’accumulation de leur toxicité avec l’âge.
Figure 4
Théorie radicalaire et autres théories du vieillissement.
L’implication des ERO dans le vieillissement n’est pas nécessairement contradictoire avec d’autres théories. Ainsi, les ERO peuvent altérer les télomères, contribuant ainsi à leur raccourcissement et à la sénescence cellulaire. Différents modèles génétiques impliquent la voie de signalisation insuline/IGF qui aboutit au facteur transcriptionnel Foxo. Or, ce facteur induit des gènes impliqués dans la résistance aux ERO et dans le stress oxydant. Par ailleurs, chez les mammifères, ce facteur est lui-même activé par les protéines Sirt, elles-mêmes impliquées dans la stabilité télomérique et la sénescence cellulaire. La restriction calorique est sans doute le modèle non génétique d’accroissement de la longévité le plus connu. La restriction calorique peut d’une part affecter la voie insuline/IGF/Foxo, et, d’autre part, le métabolisme basal et les fonctions mitochondriales, autres acteurs essentiels des théories du vieillissement. Les effets de la restriction calorique sur la consommation d’oxygène corrigée sont discutés. Néanmoins, la production d’ERO par la mitochondrie semble bien diminuer ; les protéines UCP interviendraient dans cet effet. Ainsi, la théorie radicalaire s’avère complémentaire et compatible avec d’autres visions du vieillissement.