Corps de l’article
La dysgénésie tubulaire rénale (DTR) autosomique récessive est une néphropathie rare du foetus se traduisant par une anurie précoce et persistante, responsable d’oligoamnios sévère et du cortège de malformations - dysmorphie faciale, déformation des membres et hypoplasie pulmonaire - constituant la séquence de Potter. Elle est caractérisée histologiquement par l’absence ou le nombre très réduit de tubes proximaux identifiables (Figure 1A). Depuis la description princeps par Allanson [1], le tableau clinique a été complété : un défaut d’ossification de la voûte crânienne est fréquemment présent, et une hypotension sévère et réfractaire aux traitements est signalée par quelques auteurs [2]. Des anomalies vasculaires rénales sont constamment observées. Elles sont caractérisées par un épaississement marqué de la paroi musculaire des artères préglomérulaires et interlobulaires. L’évolution de la maladie est toujours sévère, la plupart des patients mourant in utero, ou dans les 48 heures suivant la naissance, en anurie et insuffisance respiratoire. Des survies de quelques jours ou semaines ont été observées chez un très petit nombre de patients dialysés dès la ils sont restés anuriques. Seuls, à notre connaissance, deux patients ont récupéré une diurèse, mais sont en insuffisance rénale chronique ou terminale.
Figure 1
Dysgénésie tubulaire rénale.
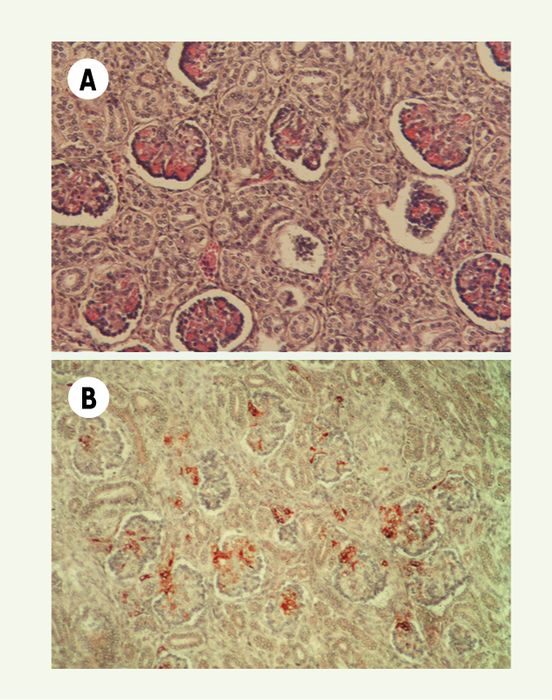
A. Absence de tubes proximaux identifiables. B . Expression rénale massive de rénine : des cellules rénine-positives sont nombreuses dans tous les appareils juxta-glomérulaires et sont également présentes dans la paroi des artères préglomérulaires et dans les axes mésangiaux
Sur la piste du système rénine-angiotensine ?
L’étiologie de ce syndrome est longtemps restée mystérieuse. Cependant, l’observation de lésions tubulaires semblables dans les reins foetaux ischémiques [3, 4] et le développement d’un phénotype clinique et pathologique similaire chez les foetus exposés à des drogues bloquant la formation ou l’action de l’angiotensine II [5, 6], situations s’accompagnant d’une hyperproduction de rénine, ont orienté nos recherches vers le système rénine-angiotensine (SRA). Ce système est constitué d’un ensemble de protéines dont l’activation aboutit à la production du peptide actif, l’angiotensine II, qui joue un rôle majeur dans la régulation de la pression artérielle et du bilan sodé. L’angiotensinogène (AGT) synthétisé par les hépatocytes, est clivé dans la circulation par la rénine, une aspartyl protéase, synthétisée et libérée par les cellules juxtaglomérulaires des artérioles afférentes du cortex rénal. L’angiotensine 1 - le décapeptide résultant de ce clivage - est convertie en angiotensine II (AII) par l’enzyme de conversion (ECA) produite par l’endothélium vasculaire et, dans le rein, par l’épithélium tubulaire proximal. L’angiotensine II se lie à deux types de récepteurs, l’AT1 médiateur de son effet vasopresseur, et l’AT2 ayant une activité antagoniste. La rénine constitue l’étape limitante de cette cascade catalytique et son taux de sécrétion dépend des conditions hémodynamiques rénales et du flux sodé dans le tube distal du rein. En outre, sa production est régulée négativement par l’AII. Tous les éléments du SRA sont exprimés dans le rein foetal humain dès la 5e semaine de gestation [7] et des travaux expérimentaux ont montré qu’ils étaient fonctionnels chez le foetus. Nous avons donc étudié l’expression rénale de la rénine, marqueur de l’activité du SRA, chez les patients atteints de DTR et réalisé une étude moléculaire des différents gènes du système chez 11 individus appartenant à 9 familles dont 5 étaient consanguines.
Découverte des gènes impliqués dans la DTR ?
Des anomalies majeures de l’expression rénale de rénine ont été observées chez tous les patients : augmentation de l’expression dans la plupart des cas (Figure 1B), absence complète de rénine dans 3 familles [8, 9]. Cette dernière observation désignait le gène REN codant la rénine comme le premier candidat pour la maladie. Effectivement, dans ces 3 familles consanguines, des mutations homozygotes du gène REN ont été identifiées. Et comme le suggérait l’absence de rénine, il s’agit de mutations « perte de fonction » : mutation non-sens dans une famille, mutations dans les sites d’épissage, décalant le cadre de lecture dans les deux autres. Le séquençage systématique de REN a permis d’identifier en outre, chez un patient d’une famille non consanguine, des mutations hétérozygotes composites : une mutation dans le site donneur d’épissage, et une mutation faux sens particulièrement intéressante car touchant l’acide aspartique 104, dans le site catalytique actif de l’enzyme, indispensable au clivage de l’angiotensinogène libérant l’angiotensine I. Cette seconde mutation permet la production de rénine, mais d’une rénine inactive, production très augmentée, par perte de la régulation négative de sa synthèse par l’AII.
Une mutation du gène AGT, codant l’angiotensinogène, a été détectée à l’état homozygote dans une famille consanguine. Cette mutation ponctuelle, du dernier nucléotide de l’exon 3, affecte le site donneur d’épissage, et entraîne théoriquement la synthèse d’une protéine ayant perdu une partie de son domaine serpine. Elle s’accompagne d’une production rénale massive de rénine, probablement par défaut d’AII, des études in vitro, ayant montré que l’intégrité du domaine serpine était nécessaire à l’interaction entre la rénine et l’angiotensinogène.
Des mutations du gène ACE ont également été identifiées : (1) délétion homozygote de 4 nucléotides, dans l’exon 8 du gène, chez 2 patients appartenant à une famille consanguine ; la protéine, si elle est produite, est non fonctionnelle car amputée de son 2e domaine catalytique, de ses séquences transmembranaire et intracytoplasmique ; (2) mutation stop dans l’exon 5, à l’état hétérozygote, dans une famille non consanguine. La rénine est intensément exprimée chez ces patients. Enfin, un foetus surexprimant la rénine a été trouvé hétérozygote composite pour le gène AGTR1 : deux mutations, une insertion d’un T et une mutation faux sens affectant une thréonine très conservée, sont présumées affecter la fonction du récepteur AT1.
Enfin, aucune mutation n’a été détectée dans une famille, suggérant que, peut-être, d’autres gènes encore pouvaient être impliqués dans cette pathologie. Les gènes codant le récepteur AT2, et le récepteur de la rénine - récemment identifiés et localisés sur le chromosome X -, n’ont pas été étudiés chez le patient de cette dernière famille puisqu’il était de sexe féminin.
Ainsi, la DTR est liée à des mutations touchant l’un ou l’autre des gènes du SRA et résultant en l’absence ou l’inefficacité de l’angiotensine II [9]. C’est la première identification de néphropathie mendélienne liée à ces gènes. Elle souligne l’importance du système dans le développement du rein foetal humain. Son mécanisme d’action reste cependant à préciser en tenant compte des fonctions multiples de l’angiotensine II : peptide vasoactif, mais également facteur de croissance tubulaire. Le développement de DTR secondaire à l’ischémie rénale [3, 4], situation qui s’accompagne d’une stimulation du SRA, suggère que c’est par le maintien d’une pression de perfusion rénale efficace que l’angiotensine II intervient dans le développement rénal. Le phénotype observé chez les patients atteints de DTR autosomique récessif est plus sévère que celui décrit chez les souris dont les différents gènes du SRA ont été invalidés. Il est très homogène, quel que soit le gène muté. Cela indique qu’il n’y a pas de redondance dans le SRA et que les voies alternes de génération de l’angiotensine II, décrites in vitro, sont inefficaces.
Parties annexes
Références
- 1. Allanson JE, Pantzar JT, MacLeod PM. Possible new autosomal recessive syndrome with unusual renal histological changes. Am J Med Genet1983 ; 16 : 57-60.
- 2. Kriegsmann J, Coerdt W, Kommos sF,et al. Renal tubular dysgenesis (RTD). An important cause of the oligo-hydramnion-sequence. Report of 3 cases and review of the literature. Pathol Res Pract2000 ; 196 : 861-5.
- 3. Landing BH, Ang SM, Herta N, et al. Labeled lectin studies of renal tubular dysgenesis and renal tubular atrophy of postnatal renal ischemia and end-stage kidney disease. Pediatr Pathol 1994 ; 14 : 87-99.
- 4. Mahieu-Caputo D, Dommergues M, Delezoide AL, et al. Twin to twin transfusion syndrome. Role of the fetal renin-angiotensin system. Am J Pathol2000 ; 156 : 629-36.
- 5. Barr M, Cohen MM. ACE inhibitor fetopathy and hypocalvaria. The kidney-skull connection. Teratology 1991 ; 44 : 485-95.
- 6. Martinovic J, Benachi A, Laurent N,et al. Fetal toxic effects of angiotensin II receptor antagonists. Report of three additional cases. Lancet 2001 ; 358 : 241-2.
- 7. Schutz S, Le Moullec JM, Corvol P, Gasc JM. Early expression of all components of the renin-angiotensin-system in human development. Am J Pathol 1996 ; 149 : 2067-79.
- 8. Gubler MC, Sarrut S, Imbert MC,et al. Renal tubular dysgenesis, an autosomal recessive disorder and the renin-angiotensin system. J Am Soc Nephrol1993 ; 4 : 263.
- 9. Gribouval O, Gonzales M, Neuhaus T, et al. Mutations in genes in the renin-angiotensin system are associated with autosomal recessive renal tubular dysgenesis. Nat Genet 2005 ; 37 : 964-8.
Liste des figures
Figure 1
Dysgénésie tubulaire rénale.
A. Absence de tubes proximaux identifiables. B . Expression rénale massive de rénine : des cellules rénine-positives sont nombreuses dans tous les appareils juxta-glomérulaires et sont également présentes dans la paroi des artères préglomérulaires et dans les axes mésangiaux