Corps de l’article
Depuis la découverte de l'implication du gène codant pour la connexine 32 (Cx32) dans la forme liée au chromosome X de la maladie de Charcot Marie-Tooth, il y a 10 ans de cela, la liste des gènes codant pour des connexines impliquées dans diverses maladies héréditaires n'a cessé de grandir. Cependant, les mécanismes physiopathologiques en jeu ne sont que partiellement connus [1].
Les obstacles dans ce domaine tiennent à la nature même des canaux formés par l'assemblage des connexines ((→) m/s 2001, n° 2, p. 244), les jonctions intercellulaires de type gap. Ces canaux permettent l'échange passif, entre deux cellules adjacentes, d'ions et de petites molécules (d'une taille inférieure a 1 kDa, cette taille pouvant varier selon l'identité de la connexine). Une jonction gap est formée par l'association de deux hémicanaux, les connexons, portés chacun par la membrane plasmique de deux cellules adjacentes. Chaque connexon résulte de l'assemblage non covalent de six connexines, identiques ou non. Ces canaux se regroupent dans la membrane pour former des plaques jonctionnelles [2].
Chez les vertébrés, plus de 20 gènes codent pour les connexines. Chacun d'entre eux a un profil d'expression propre. Les connexines possèdent quant à elles un code de compatibilité leur permettant de s'associer de façon sélective avec d'autres membres, ce qui confère au canal (homomérique ou hétéromérique) une spécificité de taille d'ouverture et une sélectivité ionique. Ainsi, l'expression de plusieurs connexines dans un même tissu permet la formation de plusieurs types de canaux, dont les propriétés sont différentes [2, 3]. Il faut également souligner le caractère extrêmement dynamique de ces canaux, dont le renouvellement dans la membrane est rapide avec, selon les tissus, des demi-vies de l'ordre de 1 à 4 heures.
Bien que la majorité des mutations pathologiques des gènes codant pour les connexines entraîne une perte de fonction, c'est-à-dire une perte de couplage intercellulaire, on ne sait pas précisément ce que les cellules échangent au travers des jonctions gap : ions, métabolites, messagers secondaires ? La découverte de mutations non associées à une perte de fonctionnalité a fortement renforcé la piste des seconds messagers : dans un article récent, M. Beltramello et al. [4] incriminent ainsi l'inositol triphosphate, Ins(1,4,5)P3, dans l'une des maladies héréditaires humaines les plus fréquentes, la surdité autosomique récessive DFNB1, due à un défaut en connexine 26 (Cx26).
Il existe un grand nombre de gènes impliqués dans des formes héréditaires de surdité ((→) m/s 2004, n° 3, p. 311).
Malgré cette hétérogénéité génétique, des mutations dans le gène codant pour la Cx26 sont responsables de plus de la moitié des cas de surdité, et ce dans la plupart des populations étudiées [5]. Deux autres gènes, codant pour Cx30 et Cx31, sont également impliqués dans des formes de surdité neurosensorielle plus rares (Tableau I).
Tableau I
Connexines et surdités.

*mutations à pénétrance incomplète.
Si les connexines Cx26 et Cx30 sont colocalisées dans la cochlée – organe de l'audition – [6], la composition précise des jonctions gap dans cet organe n'est pas précisément connue (canaux homotypiques de Cx30 ou Cx26, connexons mixtes composés des deux connexines ?). Les plaques jonctionnelles formées par ces deux molécules, particulièrement larges, définissent deux réseaux cellulaires indépendants : un réseau épithélial, qui comprend les cellules de support des cellules sensorielles et leurs cellules adjacentes, et un réseau conjonctif, composé des fibrocytes et des couches cellulaires intermédiaire et basale de la strie vasculaire (Figure 1). Cette répartition suggère que les jonctions gap joueraient un rôle clé dans l'homéostasie ionique, en participant notamment à l'évacuation rapide des ions potassiques lors de la transduction auditive.
Figure 1
Section transversale du canal cochléaire.
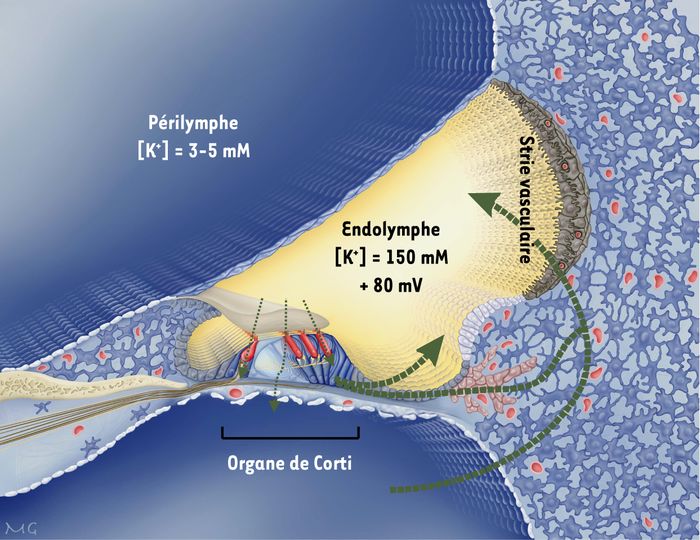
L'endolymphe est en jaune, la périlymphe en bleu. La transduction sensorielle repose sur un courant potassique dans les cellules sensorielles (en rouge). Le potassium est ensuite sécrété par ces cellules, puis recyclé. Les hypothèses de voies de recyclage sont indiquées par des flèches en pointillé.
Dans la cochlée, la surface des cellules de l'organe de Corti (épithélium sensoriel auditif) est en contact avec l'endolymphe, un liquide extracellulaire très particulier, concentré en potassium et pauvre en sodium (Figure 1). En revanche, les corps cellulaires de cet épithélium baignent dans la périlymphe, qui possède les concentrations ioniques classiques d'un milieu extracellulaire, riche en sodium et pauvre en potassium. Un autre élément clé de la transduction auditive est le potentiel électrique transépithélial, très élevé entre les compartiments périlymphatique et endolymphatique. Ce potentiel endocochléaire est essentiel, car il permet l'influx de potassium dans les cellules sensorielles lors d'une stimulation sonore, et donc leur dépolarisation. Le potassium est ensuite sécrété par les cellules sensorielles dans les espaces intercellulaires de l'organe de Corti, et en partie capté par les cellules de support [7]. Le réseau épithélial de jonctions gap permettrait d'évacuer l'excès de potassium s'accumulant autour des cellules sensorielles en conséquence d'une stimulation auditive, et éviterait à ces dernières de subir une dépolarisation prolongée et toxique. L'inactivation, chez la souris, du gène codant pour la Cx26 dans le réseau épithélial cochléaire confirme cette hypothèse [8] : chez ces animaux, l'organe de Corti se développe normalement, mais dégénère au moment de la mise en place de l'audition, lorsque le potentiel endocochléaire a atteint sa valeur adulte (environ + 80 mV). Ici, la dégénérescence cellulaire serait donc directement liée à l'activité auditive, et notamment au flux de potassium dans l'organe de Corti. Reste à expliquer pourquoi la Cx30, colocalisée avec la Cx26, ne semble pas capable de compenser l'absence de Cx26, puisque toutes les jonctions gap sont perméables au potassium [3, 9]. Cette absence de complémentation fonctionnelle est la signature des maladies liées aux connexines [1] : à chaque connexine correspond une perméabilité différente aux ions et aux messagers secondaires [10-12], ainsi qu'une sélectivité de taille et, par conséquent, une fonction physiologique donnée [2, 3]. C'est donc la combinatoire de connexines, et non la quantité de communication intercellulaire, qui prévaut. En attestent les expériences de remplacement d'une connexine par une autre réalisées chez la souris [1]. Dans ce contexte, les mutations pathologiques modifiant de façon subtile la fonction des connexines sont des outils de choix pour comprendre le rôle exact de ces molécules.
La mutation V84L du gène CX26 (GJB2) est responsable d'une surdité DFNB1, mais n'empêche pas la formation d'un canal fonctionnel [13]. M. Beltramello et al. ont donc émis l'hypothèse d'une perméabilité modifiée de ce canal : leur étude démontre que les canaux V84L ont une perméabilité réduite à l'Ins(1,4,5)P3, le messager secondaire permettant la libération de calcium des stocks intracellulaires [4]. Ainsi, dans des paires de cellules HeLa transfectées, les canaux formés par les Cx26 native ou mutée V84L ont des conductances unitaires et une probabilité d'ouverture identiques. De plus, la perméabilité de ces canaux au jaune Lucifer, un traceur fluorescent couramment utilisé pour mettre en évidence les couplages intercellulaires, est également comparable. En revanche, on observe une très nette différence de comportement des canaux V84L lors de l'injection d'Ins(1,4,5)P3 dans des cellules couplées : la cinétique et l'amplitude des changements de concentration du Ca2+ cytosolique sont alors considérablement modifiées, et la perméabilité à l'Ins(1,4,5)P3 est pratiquement abolie. La mutation V84L semble donc modifier la structure du canal, réduisant le passage d'Ins(1,4,5)P3 sans compromettre le passage des ions. La même étude rapporte également que la perméabilité des canaux Cx30 à l'Ins(1,4,5)P3 est beaucoup plus faible que celle des canaux Cx26 : la Cx30 ne serait donc pas capable de remplacer la Cx26.
M. Beltramello et al. proposent donc que la présence de la forme V84L de la Cx26 réduit la perméabilité de l'Ins(1,4,5)P3 dans l'organe de Corti [4]. Cet effet peut-il être responsable de surdité ? Afin d'ébaucher une réponse à cette question cruciale, M. Beltramello et al. ont utilisé des cultures organotypiques d'organe de Corti de rat pour étudier la diffusion d'Ins(1,4,5)P3. Leur étude montre que la diffusion, au travers des jonctions gap, d'Ins(1,4,5)P3 injecté dans une cellule de support induit la propagation de vagues calciques, également dépendantes de la voie purinergique activée par la sécrétion d'ATP dans le milieu extracellulaire [4]. Des études précédentes avaient montré qu'une élévation de la concentration de Ca2+ cytosolique dans les cellules de support active un flux sortant de potassium et de chlore dans l'endolymphe [14]. Les résultats de M. Beltramello et al. suggèrent que la présence de jonctions gap V84L Cx26 modifierait l'homéostasie potassique de l'endolymphe (Figure 2).
Figure 2
Modèle hypothétique du rôle proposé pour la diffusion d'Ins(1,4,5)P3 dans la transduction auditive.
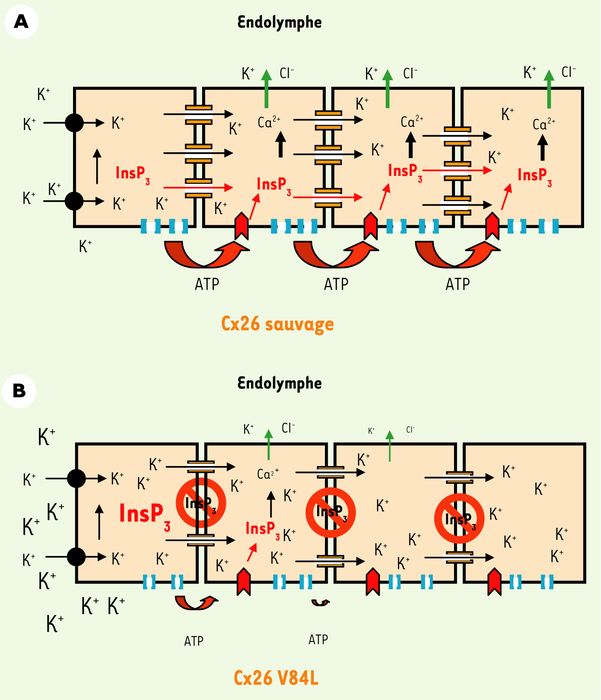
Lors d'une stimulation sonore, le potassium est sécrété par les cellules sensorielles dans les espaces intercellulaires de l'organe de Corti, puis capté par les cellules de support via un cotransporteur K+/Cl-, Kcc4 (cercle noir). Les jonctions gap permettraient d'éliminer l'excès de potassium s'accumulant autour des cellules sensorielles (flèches noires). A. La Cx26 sauvage (en orange) permet également le passage intercellulaire d'Ins(1,4,5)P3 (flèches rouges), assurant la propagation de vagues calciques. Le stimulus responsable de l'augmentation initiale d'Ins(1,4,5)P3 n'est pas compris, mais cette étape induirait l'activation d'un système d'extrusion K+/Cl- (flèches vertes) contribuant au recyclage du potassium dans l'endolymphe. Les vagues intercellulaires de Ca2+ sont également dépendantes de l'activation de récepteurs purinergiques (en rouge), par une voie paracrine impliquant probablement des hémicanaux de connexines (bleu clair), ou un autre mécanisme. B. La diffusion d'Ins(1,4,5)P3 à travers les canaux V84L (Cx26 mutée) est fortement diminuée (signaux d'interdiction), sans que la perméabilité ionique soit affectée (flèches noires). Il en résulterait une diminution de l'activité d'extrusion K+/Cl- et l'accumulation d'ions potassiques dans l'espace extracellulaire autour des cellules sensorielles et de support, entraînant une dépolarisation prolongée et toxique de ces cellules.
Restent de nombreuses questions sans réponse. Par exemple, les vagues calciques observées lors de l'application d'ATP à la surface de l'organe de Corti, ou au cours d'une stimulation mécanique, ont-elles réellement lieu lors de la transduction auditive ? L'imagerie calcique in vivo devrait permettre de répondre à cette question. Et, si le modèle proposé ici est exact, on devrait constater une diminution de la propagation de ces vagues dans le modèle murin où la Cx26 n'est plus exprimée dans l'organe de Corti, ou dans un modèle où la Cx26 serait remplacée par la forme mutée V84L.
Pour conclure, la tentation est grande d'incriminer la diffusion de messagers secondaires au travers des jonctions gap dans d'autres maladies. Cette idée n'est pas nouvelle : le transfert d'AMPc au travers de canaux formés par la Cx32 avait été indirectement mis en cause dans la forme liée au chromosome X de la maladie de Charcot-Marie-Tooth [15]. D'autres exemples suivront sans doute.
Parties annexes
Références
- 1. Gerido DA, White TW. Connexin disorders of the ear, skin, and lens. Biochim Biophys Acta 2004 ; 1662 : 159-70.
- 2. Bruzzone R, White TW, Paul, DL. Connections with connexins : the molecular basis of direct intercellular signaling. Eur J Biochem 1996 ; 238 : 1-27.
- 3. Harris AL. Emerging issues of connexin channels : biophysics fills the gap. Q Rev Biophys 2001 ; 34 : 325-472.
- 4. Beltramello M, Piazza V, Bukauskas FF, et al. Impaired permeability to Ins(1,4,5)P3 in a mutant connexin underlies recessive hereditary deafness. Nat Cell Biol 2005 ; 7 : 63-9.
- 5. Petit C, Levilliers J, Hardelin JP. Molecular genetics of hearing loss. Annu Rev Genet 2001 ; 35 : 589-646.
- 6. Lautermann J, Ten Cate WJ, Altenhoff P, et al. Expression of the gap-junction connexins 26 and 30 in the rat cochlea. Cell Tissue Res 1998 ; 294 : 415-20.
- 7. Boettger T, Hubner CA, Maier H, et al. Deafness and renal tubular acidosis in mice lacking the K-Cl co-transporter Kcc4. Nature 2002 ; 416 : 874-8.
- 8. Cohen-Salmon M, Ott T, Michel V, et al. Targeted ablation of connexin26 in the inner ear epithelial gap junction network causes hearing impairment and cell death. Curr Biol 2002 ; 12 : 1106-11.
- 9. Valiunas V, Manthey D, Vogel R, et al. Biophysical properties of mouse connexin30 gap junction channels studied in transfected human HeLa cells. J Physiol 1999 ; 519 : 631-44.
- 10. Bevans CG, Kordel M, Rhee SK, Harris AL. Isoform composition of connexin channels determines selectivity among second messengers and uncharged molecules. J Biol Chem 1998 ; 273 : 2808-16.
- 11. Goldberg GS, Lampe PD, Nicholson BJ. Selective transfer of endogenous metabolites through gap junctions composed of different connexins. Nat Cell Biol 1999 ; 1 : 457-9.
- 12. Niessen H, Harz H, Bedner P, et al. Selective permeability of different connexin channels to the second messenger inositol 1,4,5-trisphosphate. J Cell Sci 2000 ; 113 : 1365-72.
- 13. Bruzzone R, Gomès D, Veronesi V, et al. Functional analysis of recessive mutations of human connexin26 associated with nonsyndromic deafness. FEBS Lett 2003 ; 533 : 79-88.
- 14. Gale JE, Piazza V, Ciubotaru CD, Mammano F. A mechanism for sensing noise damage in the inner ear. Curr Biol 2004 ; 14 : 526-9.
- 15. Oh S, Ri Y, Bennett MV, et al. Changes in permeability caused by connexin 32 mutations underlie X-linked Charcot-Marie-Tooth disease. Neuron 1997 ; 19 : 927-38.
Liste des figures
Figure 1
Section transversale du canal cochléaire.
L'endolymphe est en jaune, la périlymphe en bleu. La transduction sensorielle repose sur un courant potassique dans les cellules sensorielles (en rouge). Le potassium est ensuite sécrété par ces cellules, puis recyclé. Les hypothèses de voies de recyclage sont indiquées par des flèches en pointillé.
Figure 2
Modèle hypothétique du rôle proposé pour la diffusion d'Ins(1,4,5)P3 dans la transduction auditive.
Lors d'une stimulation sonore, le potassium est sécrété par les cellules sensorielles dans les espaces intercellulaires de l'organe de Corti, puis capté par les cellules de support via un cotransporteur K+/Cl-, Kcc4 (cercle noir). Les jonctions gap permettraient d'éliminer l'excès de potassium s'accumulant autour des cellules sensorielles (flèches noires). A. La Cx26 sauvage (en orange) permet également le passage intercellulaire d'Ins(1,4,5)P3 (flèches rouges), assurant la propagation de vagues calciques. Le stimulus responsable de l'augmentation initiale d'Ins(1,4,5)P3 n'est pas compris, mais cette étape induirait l'activation d'un système d'extrusion K+/Cl- (flèches vertes) contribuant au recyclage du potassium dans l'endolymphe. Les vagues intercellulaires de Ca2+ sont également dépendantes de l'activation de récepteurs purinergiques (en rouge), par une voie paracrine impliquant probablement des hémicanaux de connexines (bleu clair), ou un autre mécanisme. B. La diffusion d'Ins(1,4,5)P3 à travers les canaux V84L (Cx26 mutée) est fortement diminuée (signaux d'interdiction), sans que la perméabilité ionique soit affectée (flèches noires). Il en résulterait une diminution de l'activité d'extrusion K+/Cl- et l'accumulation d'ions potassiques dans l'espace extracellulaire autour des cellules sensorielles et de support, entraînant une dépolarisation prolongée et toxique de ces cellules.
Liste des tableaux
Tableau I
Connexines et surdités.
*mutations à pénétrance incomplète.