Résumés
Résumé
Les puces à ADN sont des multicapteurs permettant de caractériser et quantifier un acide nucléique dans un échantillon. Elles apportent une solution innovante au problème ancien de la détection, de l’identification et du typage de bactéries dans un échantillon. Elles permettent la caractérisation génomique rapide de bactéries pathogènes et facilitent les études épidémiologiques, par exemple pour le contrôle des maladies nosocomiales ou la surveillance du bioterrorisme. Ces puces sont développées dans les laboratoires de recherche pour l’étude de la diversité et de l’évolution du monde bactérien, pour rechercher des gènes de résistance aux antibiotiques et pour la caractérisation de communautés bactériennes constituées de centaines d’espèces. L’industrialisation du processus de fabrication et d’utilisation, rendant la technologie robuste tout en en diminuant son coût, devrait permettre son utilisation dans les laboratoires hospitaliers et d’analyses spécialisées, puis sa généralisation aux laboratoires de ville.
Summary
DNA-arrays are mainly known for their application in transcriptome analysis leading for instance to the discovery of new marker genes for diagnostics and prognostics in oncology. However, DNA arrays are also used for massively parallel analysis of DNA molecules allowing their quantification, the detection of single nucleotide polymorphisms and re-sequencing. This multi detection system is now applied to the « old » problems of detecting and identifying bacteria in a biological sample and for the fine molecular characterization of a bacterial isolate. This new tool should serve for the diagnostic of an infection and for epidemiological studies such as those performed for the control of nosocomial infections or for the surveillance of bioterrorism attacks. DNA arrays carrying probes for 16S RNA specific of hundreds of bacterial species allow the identification of bacteria within a community by a single hybridization of amplified 16S rDNAs with universal primers and re-sequencing DNA arrays are used for multi locus sequence typing in a single step. Finally, the genome of an isolate could be characterized by DNA-arrays focused on a specific question like presence of toxin or antibiotic resistance genes. Up to now, DNA arrays are used in research laboratories for the rapid characterization at the genomic level of a strain collection, for evolutionary and population genetics studies and for the characterization of bacterial communities. Industrializing the process of DNA-array construction and hybridization is now needed in order to transfer this technology to hospitals and diagnostic laboratories.
Corps de l’article
Depuis la démonstration par Koch, en 1876, que la maladie du charbon est due à Bacillus anthracis [1], l’identification des microbes responsables des maladies infectieuses a été une priorité de la microbiologie clinique. L’isolement du germe et sa description phénotypique sont nécessaires pour mieux comprendre la maladie qu’il provoque et pour un diagnostic sûr. L’épidémiologie des maladies infectieuses cherche à élucider les mécanismes de transmission des agents infectieux, l’existence de réservoirs et leur origine. L’identification de l’espèce n’est alors pas suffisante et la caractérisation plus fine (le typage) de la bactérie isolée est nécessaire afin de déterminer la probabilité pour deux isolats d’avoir la même origine. Répondre à cette question a des implications multiples : pour les maladies nosocomiales, ces recherches doivent permettre d’identifier l’origine hospitalière de l’infection et de mettre en place les procédures pour y remédier [2] ; pour des maladies d’origine alimentaire ou environnementale, comme la listériose ou la légionellose, des réseaux de surveillance[1] cherchent à identifier par des études épidémiologiques la source de la contamination, afin de l’éliminer. La démonstration de l’origine d’une infection a et aura de plus en plus des implications légales, et le typage des bactéries incriminées est un des éléments de l’action judiciaire. La crainte du bioterrorisme pousse également à rechercher des outils performants pour identifier les germes dispersés dans l’environnement, pour retrouver l’origine des souches et comprendre les éventuels trafics de ces armes biologiques.
Des méthodes de microbiologie classique développées au xixe siècle comme la coloration de Gram aux analyses moléculaires récentes fondées sur l’analyse des acides nucléiques, les microbiologistes ont développé de très nombreuses méthodes d’identification et de typage des bactéries, mettant à profit leur diversité [3]. L’objectif de cet article est de montrer le formidable potentiel des puces à ADN dans ce domaine, mais aussi d’analyser les raisons pour lesquelles cette technologie tarde à s’implanter dans les laboratoires d’analyse microbiologique.
La diversité du monde bactérien
La caractérisation de la diversité bactérienne sous-tend toutes les méthodes d’identification. Au cours de la dernière décennie, notre conception de cette diversité a été transformée par l’analyse des génomes. Au mois d’août 1995, la communauté scientifique a été surprise par la publication de la séquence du génome d’Haemophilus influenzae [4]. La séquence du génome d’une bactérie modèle était attendue, mais c’est une bactérie d’importance clinique, peu étudiée, pour laquelle n’étaient disponibles ni carte physique ni carte génétique, qui a vu la première l’ensemble de ses gènes décryptés. La démonstration que le séquençage d’un génome bactérien pouvait être réalisé rapidement a ouvert la porte à la systématisation de son application aux bactéries pathogènes, qui représentent la majorité des plus de 180 séquences génomiques publiées à ce jour[2]. La connaissance du génome permet le développement de nouvelles méthodes de typage moléculaire, mais aussi phénotypique, par la découverte d’activités enzymatiques spécifiques.
Cependant, le décryptage du génome d’un isolat n’est pas suffisant pour connaître une espèce. La disponibilité de séquences génomiques de plusieurs souches a permis de mettre en évidence la diversité génomique d’une espèce, non seulement au niveau du polymorphisme de chaque gène, mais aussi du répertoire de gènes présents. Il est possible de distinguer dans un génome un squelette conservé entre tous les isolats d’une espèce et un ensemble d’îlots qui sont spécifiques d’un clone ou d’un lignage particulier [5]. Cette partie variable du génome est constituée d’éléments mobiles comme des bactériophages, des plasmides ou des transposons, mais également de groupes de gènes insérés ou délétés indépendamment de tout système de recombinaison spécifique. Ces gènes apportent des fonctions particulières à un clone, comme des fonctions métaboliques, de virulence, de production de toxines ou de résistance à des antibiotiques. Cette diversité génomique explique aussi la diversité de virulence retrouvée au sein d’une espèce. Les méthodes de typage moléculaire sont fondées sur le polymorphisme des séquences et la diversité du contenu génétique des génomes ; les puces à ADN permettent d’analyser ces deux aspects.
Méthodes d’identification et de typage
L’identification bactérienne nécessite l’isolement de la bactérie sous forme d’une colonie. Cette première étape peut être difficile si l’échantillon biologique n’est pas normalement stérile, comme un prélèvement de gorge ou un prélèvement de selles. Traditionnellement, l’identification de l’espèce se faisait en combinant l’observation microscopique et l’analyse phénotypique, en étudiant la forme et la couleur des colonies et les propriétés métaboliques et enzymatiques. Cette méthode peut être remplacée par une analyse moléculaire soit en utilisant des tests immunologiques, soit sur la base de séquences d’ADN, par exemple après amplification par PCR. Les méthodes de PCR présentent l’avantage de pouvoir être réalisées directement à partir d’un échantillon biologique. Le gène codant pour l’ARN ribosomique 16S est un des marqueurs d’espèce les plus utilisés : il est en effet possible de définir des amorces universelles pour son amplification, et la comparaison de sa séquence avec des banques de données de référence permet de déterminer l’espèce [6].
De la même manière, après l’identification de l’espèce, les premières méthodes de typage bactérien étaient phénotypiques, par comparaison des antigènes de surface (sérotypie) ou analyse de la sensibilité à des bactériophages (lysotypie). Durant ces vingt dernières années, de nombreuses méthodes de typage moléculaire ont été développées [7]. La valeur de ces méthodes dépend de leur pouvoir de discrimination entre les isolats, de leurs facilité et rapidité d’utilisation et de la reproductibilité des résultats entre différents laboratoires. Cette reproductibilité est essentielle pour l’échange des données, leur organisation sous forme de bases de données internationales et la mise en place de systèmes de surveillance épidémiologique performants. Les méthodes considérées aujourd’hui comme les plus fiables sont l’analyse des fragments de restriction de l’ADN chromosomique après migration en champ pulsé (pulsotype) et l’analyse des séquences nucléotidiques de plusieurs gènes de ménage (exprimés dans toutes les cellules) (STML, séquençotypage multilocus, MLST en anglais). Le pulsotype dépend du polymorphisme des sites de reconnaissance par les enzymes de restriction et de l’organisation du génome. Le type MLST dépend uniquement de la vitesse d’évolution nucléotidique au sein de l’espèce et d’éventuels transferts génétiques horizontaux.
Puces à ADN
Le principe d’une puce à ADN réside dans la reconnaissance, c’est-à-dire l’hybridation, entre deux molécules d’ADN simple brin complémentaires (Figure 1). L’échantillon (ADN ou ARN), marqué de manière fluorescente, est mis en contact avec la puce portant plusieurs milliers de sondes qui sont des fragments d’ADN ou des oligonucléotides de séquence connue [8, 9]. Après lavage du matériel fixé de manière non spécifique, le signal est quantifié au niveau de chaque sonde. Sa valeur dépendra de la concentration en molécules marquées complémentaires de la sonde dans l’échantillon et du degré de complémentarité (le pourcentage d’identité) avec la sonde.
Figure 1
Analyse d’acide nucléique par puce à ADN.
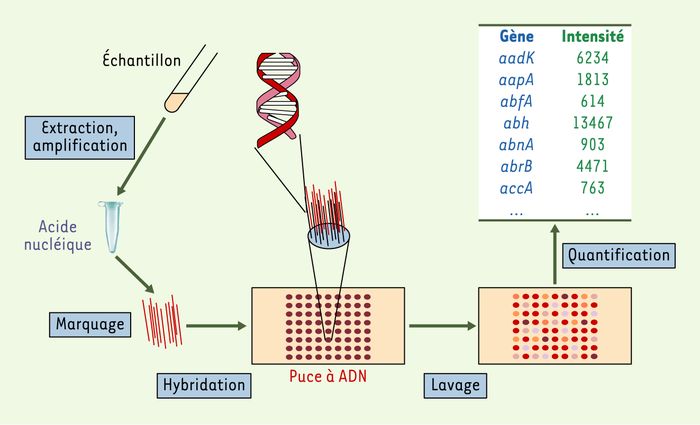
L’ADN ou l’ARN est purifié et éventuellement amplifié à partir d’un échantillon biologique. Il est ensuite marqué de manière fluorescente et mis en contact avec les sondes portées par la puce. Lors de cette étape d’hybridation, les acides nucléiques marqués vont s’apparier avec les sondes ADN fixées sur le support. Une étape de lavage permet ensuite d’éliminer les acides nucléiques marqués fixés de manière non spécifique. Finalement, la fluorescence au niveau de chaque dépôt de sonde sera quantifiée au moyen de tubes photomultiplicateurs ou d’une caméra CCD (charge-coupled device). Les valeurs obtenues pour chaque sonde, comme indiquées sur le tableau, doivent ensuite être traitées aux moyens d’outils informatiques pour obtenir la caractérisation de l’échantillon.
Actuellement, l’utilisation la plus courante des puces à ADN concerne la quantification des ARN messagers d’un échantillon (transcriptome) afin de comparer les profils de transcription de deux échantillons obtenus dans des conditions de croissance différentes. Dans le cadre de l’identification bactérienne et du typage, leur utilisation est différente : ces puces permettent la détection d’une séquence d’ADN dans un mélange, d’identifier un polymorphisme de séquence (SNP) et de re-séquencer un fragment d’ADN (Figure 2).
Figure 2
Trois applications des puces à ADN dans l’analyse microbiologique.

A. Analyse d’une population bactérienne mixte. Après extraction de l’ADN, les régions codant pour l’ARN ribosomique 16S sont amplifiées pour toutes les bactéries de l’échantillon en utilisant des amorces universelles correspondant à des régions conservées dans toutes les espèces. La puce porte des oligonucléotides de séquence spécifique de chaque espèce. Après hybridation, la quantification du signal permet de détecter la présence de bactéries des différentes espèces représentées sur la puce et d’évaluer leurs quantités relatives. B. Puce Affymetrix de re-séquençage (http://www.affymetrix.com/). Ces puces portent plusieurs centaines de milliers d’oligonucléotides synthétisés in situ par une technique de photolithographie. Pour chaque base, quatre oligonucléotides sont synthétisés d’après une séquence de référence avec à la position centrale l’une des quatre bases, A, C, G ou T. La comparaison des signaux de fluorescence pour ces quatre oligonucléotides permet de déterminer la séquence à cette position. C. Détection de régions chromosomiques spécifiques d’un isolat. L’ADN de la souche à analyser et un ADN de référence sont marqués par des fluorophores ayant des propriétés spectrales différentes. La puce à ADN est hybridée avec un mélange des deux ADN marqués. Après analyse aux deux longueurs d’onde d’émission de fluorescence, les sondes absentes de la souche analysée n’émettront que pour la longueur d’onde du fluorophore de l’ADN de référence.
Les puces à ADN comme outils de détection
Des puces à ADN sont utilisées pour détecter un produit de PCR et remplacent ainsi la migration sur gel en validant la spécificité de l’amplification par sa complémentarité avec la sonde. Pour l’analyse d’un échantillon simple, cette procédure est peu compétitive par rapport à la PCR en temps réel ou à l’électrophorèse capillaire. En revanche, dans le cas d’un échantillon complexe, les puces à ADN permettent l’analyse simultanée d’un grand nombre de fragments. Des puces permettant d’identifier les bactéries présentes dans un échantillon sur la base de l’analyse des séquences d’ARN ribosomique 16S sont en développement dans plusieurs institutions (Figure 2A). L’ADN total est extrait à partir d’un échantillon et l’ensemble des ADN codant pour les ARN 16S sont amplifiés, en utilisant des amorces universelles, et hybridés sur des puces portant des sondes spécifiques de l’ARN 16S des espèces recherchées. Cet outil est développé pour la surveillance du bioterrorisme avec un ensemble de sondes spécifiques des agents pathogènes potentiellement utilisés.
Les puces à ADN pour le re-séquençage
La connaissance de la séquence complète d’un génome est le niveau ultime de typage, mais, dans le contexte actuel, le séquençage de chaque isolat n’est pas envisageable. Des puces oligonucléotides sont donc développées pour obtenir des informations partielles sur les génomes bactériens (Figure 2B). Des puces sont utilisées pour le typage par MLST de Staphylococcus aureus [10]. Les sept locus analysés sont amplifiés par PCR et hybridés à la puce au lieu d’être séquencés un par un. La société BioMérieux, en collaboration avec Affymetrix, a été pionnière dans le domaine en développant des puces pour l’identification de Mycobacterium tuberculosis et la recherche de mutations entraînant la résistance à l’isoniazide et à la rifampicine [11]. L’amélioration de la sensibilité de la technique doit permettre d’utiliser l’ADN génomique total pour détecter un ensemble de positions polymorphes réparties sur le génome. Il serait alors possible d’avoir une vision globale du génome et de pointer sur des mutations particulières, comme celles entraînant la résistance à un antibiotique.
Les puces à ADN pour la caractérisation génomique
Les puces à ADN sont aussi utilisées pour caractériser la partie variable du génome d’un clone. Ces puces « biodiversité » portent des sondes correspondant à des gènes qui ne sont pas présents dans tous les isolats d’une espèce (Figure 2C). Elles sont établies à partir de la comparaison des séquences de plusieurs génomes. Par une seule expérience d’hybridation, ces puces permettent d’établir une véritable empreinte digitale correspondant aux gènes présents ou absents dans un clone. Ainsi, à la différence de la majorité des méthodes de typage, les puces à ADN apportent une information fonctionnelle sur la nature des gènes qui différencient deux isolats. Ces résultats peuvent être comparés aux données phénotypiques sur les souches, notamment en relation avec leur virulence. De telles puces de typage ont été établies pour Listeria monocytogenes, sur la base de la séquence génomique de deux isolats et d’un isolat de L. innocua, une espèce non pathogène très proche de L. monocytogenes [12], et pour S. aureus, en combinant les connaissances de sept génomes et en ajoutant des gènes de résistance aux antibiotiques ainsi que des gènes codant pour des toxines [13]. Dans les deux cas, l’analyse d’une collection de souches par hybridation de ces puces à ADN a montré qu’elles étaient un outil de typage puissant, aussi résolutif que l’analyse en champ pulsé. Par ailleurs, pour S. aureus, ces puces se sont révélées extrêmement efficaces pour l’identification des résistances aux antibiotiques, avec un très bon accord entre les résultats d’hybridation et les résultats d’antibiogramme. Finalement, la confrontation des données de phylogénie, fondée sur une analyse par MLST et la distribution des gènes entre les isolats, permet de mettre en évidence des phénomènes de transferts génétiques horizontaux qui peuvent jouer un rôle important dans l’émergence de clones hypervirulents.
Transfert vers les laboratoires d’analyse
Les résultats obtenus dans les laboratoires de recherche montrent l’apport des puces à ADN en épidémiologie moléculaire et leur potentiel pour de nombreuses recherches en microbiologie. Théoriquement, c’est une technologie qui peut facilement être automatisée et industrialisée. Pourtant, alors que l’utilisation des puces Affymetrix pour M. tuberculosis a été publiée en 1997, cette technologie reste très confidentielle et il n’existe pas de produit de typage, fondé sur les puces à ADN, commercialisé à grande échelle. La première raison à ce délai est technologique. Les méthodes développées sont encore contraignantes et l’obtention de résultats reproductibles nécessite un ADN génomique d’une grande qualité. Des améliorations techniques accompagnées d’une simplification de la méthode augmentant sa robustesse sont nécessaires. La seconde raison à ce délai est liée à la difficulté de modifier des procédures reconnues au niveau international. Il est nécessaire qu’une nouvelle méthode soit d’abord validée et utilisée par les centres nationaux de référence et démontre sa supériorité avant de voir son utilisation systématisée dans les laboratoires d’analyse. La mise en place de sites Web équivalents à ceux qui ont été développés pour le MLST[3] devrait aussi promouvoir cette méthodologie. Les approches génomiques d’identification présentent un potentiel d’applications multiples et il est très probable que les problèmes technologiques seront résolus avec l’industrialisation du processus, en s’accompagnant d’une baisse des coûts. L’identification bactérienne devrait profiter des développements liés à l’utilisation des puces dans d’autres domaines de la santé, comme l’analyse des tumeurs ou de la prédisposition à certaines maladies. En retour, les développements réalisés en microbiologie clinique auront des répercussions en microbiologie de l’environnement et en microbiologie alimentaire.
Conclusions et perspectives
Les progrès de la génomique et la prise en compte de la génétique des populations pour les bactéries pathogènes permettent de mieux comprendre l’évolution et la diversité au sein des espèces pathogènes. La combinaison d’un outil performant de caractérisation des isolats bactériens et de bases de données internationales ouvertes incluant des isolats d’origines très diverses est l’occasion d’établir de nouveaux liens entre la surveillance microbiologique, la recherche clinique et la recherche fondamentale. Outre la caractérisation des génomes bactériens, l’application des puces à ADN pour l’analyse du transcriptome a contribué de manière très significative aux progrès récents dans l’étude du processus infectieux par une meilleure compréhension de la réponse de l’hôte et du parasite et des communications qui s’instaurent entre les deux partenaires au cours de la maladie [14].
Parties annexes
Notes
-
[1]
Voir par exemple l’organisation EWGLI pour la légionellose (http://www.ewgli.org/), le réseau européen med-vet-net pour la prévention des zoonoses (http://www.medvetnet.org) ou, aux États-Unis, le réseau Pulsenet pour les infections d’origine alimentaire (http://www.cdc.gov/pulsenet).
- [2]
- [3]
Références
- 1. Koch R. Die Aetiologie der Milzbrand-Krankheit, begründet auf die Entwicklungsgeschichte des Bacillus anthracis.Beiträge zur Biologie der Pflanzen 1876 ; 1 : 277-308.
- 2. Struelens M. Molecular typing : a key tool for the surveillance and control of nosocomial infection. Curr Opin Infect Dis 2002 ; 15 : 383-5.
- 3. Raoult D, Fournier PE, Drancourt M. What does the future hold for clinical microbiology ? Nat Rev Microbiol 2004 ; 2 : 151-9.
- 4. Fleischmann RD, Adams MD, White O, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 1995 ; 269 : 496-512.
- 5. Lan R, Reeves PR. Intraspecies variation in bacterial genomes : the need for a species genome concept. Trends Microbiol 2000 ; 8 : 396-401.
- 6. Stackebrandt E, Goebel BM. Taxonomic note : a place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int J Syst Bacteriol 1994 ; 44 : 846-9.
- 7. Van Belkum A. High-throughput epidemiologic typing in clinical microbiology. Clin Microbiol Infect 2003 ; 9 : 86-100.
- 8. Schena M, Shalon D, Heller R, et al. Parallel human genome analysis : microarray-based expression monitoring of 1000 genes. Proc Natl Acad Sci USA 1996 ; 93 : 10614-9.
- 9. Southern EM. DNA chips : analysing sequence by hybridization to oligonucleotides on a large scale. Trends Genet 1996 ; 12 : 110-5.
- 10. Van Leeuwen WB, Jay C, Snijders S, et al. Multilocus sequence typing of Staphylococcus aureus with DNA array technology. J Clin Microbiol 2003 ; 41 : 3323-6.
- 11. Troesch A, Nguyen H, Miyada CG, et al.Mycobacterium species identification and rifampin resistance testing with high-density DNA probe arrays. J Clin Microbiol 1999 ; 37 : 49-55.
- 12. Doumith M, Cazalet C, Simoes N, et al. New aspects regarding evolution and virulence of Listeria monocytogenes revealed by comparative genomics. Infect Immun 2004 ; 72 : 1072-83.
- 13. Trad S, Allignet J, Frangeul L, et al. DNA macroarray for identification and typing of Staphylococcus aureus isolates. J Clin Microbiol 2004 ; 42 : 2054-64.
- 14. Bryant PA, Venter D, Robins-Browne R, Curtis N. Chips with everything : DNA microarrays in infectious diseases. Lancet Infect Dis 2004 ; 4 : 100-11.
Liste des figures
Figure 1
Analyse d’acide nucléique par puce à ADN.
L’ADN ou l’ARN est purifié et éventuellement amplifié à partir d’un échantillon biologique. Il est ensuite marqué de manière fluorescente et mis en contact avec les sondes portées par la puce. Lors de cette étape d’hybridation, les acides nucléiques marqués vont s’apparier avec les sondes ADN fixées sur le support. Une étape de lavage permet ensuite d’éliminer les acides nucléiques marqués fixés de manière non spécifique. Finalement, la fluorescence au niveau de chaque dépôt de sonde sera quantifiée au moyen de tubes photomultiplicateurs ou d’une caméra CCD (charge-coupled device). Les valeurs obtenues pour chaque sonde, comme indiquées sur le tableau, doivent ensuite être traitées aux moyens d’outils informatiques pour obtenir la caractérisation de l’échantillon.
Figure 2
Trois applications des puces à ADN dans l’analyse microbiologique.
A. Analyse d’une population bactérienne mixte. Après extraction de l’ADN, les régions codant pour l’ARN ribosomique 16S sont amplifiées pour toutes les bactéries de l’échantillon en utilisant des amorces universelles correspondant à des régions conservées dans toutes les espèces. La puce porte des oligonucléotides de séquence spécifique de chaque espèce. Après hybridation, la quantification du signal permet de détecter la présence de bactéries des différentes espèces représentées sur la puce et d’évaluer leurs quantités relatives. B. Puce Affymetrix de re-séquençage (http://www.affymetrix.com/). Ces puces portent plusieurs centaines de milliers d’oligonucléotides synthétisés in situ par une technique de photolithographie. Pour chaque base, quatre oligonucléotides sont synthétisés d’après une séquence de référence avec à la position centrale l’une des quatre bases, A, C, G ou T. La comparaison des signaux de fluorescence pour ces quatre oligonucléotides permet de déterminer la séquence à cette position. C. Détection de régions chromosomiques spécifiques d’un isolat. L’ADN de la souche à analyser et un ADN de référence sont marqués par des fluorophores ayant des propriétés spectrales différentes. La puce à ADN est hybridée avec un mélange des deux ADN marqués. Après analyse aux deux longueurs d’onde d’émission de fluorescence, les sondes absentes de la souche analysée n’émettront que pour la longueur d’onde du fluorophore de l’ADN de référence.