Résumés
Résumé
Le système rénine-angiotensine, l’un des principaux complexes de régulation de la pression sanguine, est distribué entre le sang circulant et l’espace péricellulaire de l’interstium tissulaire. Il participe en physiologie et en pathologie de la régulation de la vasomotricité et du remodelage tissulaire dans le système cardiovasculaire. Dans le cadre de ces effets, le système rénine-angiotensine tissulaire agit sur les cellules musculaires lisses vasculaires et les fibroblastes, tandis que le système rénine-angiotensine plasmatique a pour cibles les cellules endothéliales et les leucocytes circulants. L’angiotensine II, peptide actif du système, déclenche différentes voies de signalisation aboutissant à une réponse fonctionnelle immédiate (hypertension artérielle), puis à une réponse structurale hypertrophiante et, enfin, à des réponses pro-inflammatoires et procoagulantes. Dans des modèles expérimentaux d’athérosclérose, la perfusion d’angiotensine II induit la formation d’anévrismes, qui a été reliée à l’activation des leucocytes circulants. Des antagonistes de l’angiotensine II ont, dans ce type de modèle, un effet bénéfique sur le ralentissement de la formation des lésions d’athérosclérose.
Summary
The renin-angiotensin system (RAS) is compartmented between circulating blood and tissue pericellular space. Whereas renin and its substrate diffuse easily from one compartment to another, the angiotensin peptides act in the compartment where there are generated: blood or pericellular space. Renin is trapped in tissues by low and high affinity receptors. In the target cells, angiotensin II/AT1 receptor interaction generates different signals including an immediate functional calcium-dependent response, secondary hypertrophy and a late proinflammatory and procoagulant response. These late pathological effects are mediated by NADPH oxydase-generated free oxygen radicals and NFκB activation. In vivo, the tissue binding of renin and the induction of converting enzyme are the main determinants of the involvement of the RAS in vascular remodeling. The target cells of interstitial angiotensin II are mainly the vascular smooth muscle cells and fibroblasts, whereas the endothelial cells and circulating leukocytes are the main targets of circulating angiotensin II. In vivo, angiotensin II participates in the vascular wall hypertrophy associated with hypertension. In diabetes, as in other localized fibrotic cardiovascular diseases, the tissue effects of angiotensin II are mainly dependent on its ability to induce TGF-β expression. In experimental atherosclerosis, angiotensin II infusion induces aneurysm formation mediated by activation of circulating leucocytes. In these models, the administration of angiotensin II antagonists has beneficial effects on pathological remodeling. Such beneficial effects of angiotensin II antagonists in localized pathological remodeling have not yet been demonstrated in humans.
Corps de l’article
Le système rénine-angiotensine (SRA) consiste en une cascade d’interactions biochimiques aboutissant à la production de l’angiotensine II, un peptide de huit acides aminés qui interagit avec ses cellules cibles via des récepteurs à sept domaines transmembranaires couplés à des phospholipases par l’intermédiaire de protéines G. Nous nous intéresserons ici aux cibles directes de la rénine et de l’angiotensine II dans le système vasculaire. Une autre action importante de l’angiotensine II est de stimuler la sécrétion d’aldostérone par la corticosurrénale (les effets de l’aldostérone sur le remodelage vasculaire ne seront toutefois pas abordés ici, car ils nécessitent qu’un article entier leur soit consacré).
Compartimentation physiologique du système rénine-angiotensine
Le système rénine-angiotensine a été décrit comme un système endocrine par H. Goldblatt en 1934 [1].
La rénine active est synthétisée et stockée par les cellules myoépithélioïdes de l’artériole afférente du glomérule rénal. Sa sécrétion est contrôlée par divers stimulus diminuant la concentration de calcium libre dans la cellule myoépithélioïde : stimulation β-adrénergique, baisse de la tension pariétale dans l’artériole et diminution de la réabsorption du sodium et du chlore par le cotransport Na-K-2Cl de la macula densa. Inversement, l’augmentation du NaCl dans la macula densa, de même que l’angiotensine II elle-même, freinent la sécrétion de rénine en augmentant la concentration de calcium libre dans les cellules myoépithélioïdes.
La rénine active sécrétée diffuse dans les compartiments plasmatique, lymphatique et interstitiel, de même que son substrat, l’angiotensinogène, synthétisé et sécrété par le foie (il est également probable que de faibles quantités d’angiotensinogène soient directement synthétisées au niveau de la paroi artérielle). À l’opposé, les peptides dérivés de l’angiotensinogène (angiotensines I et II) n’agissent vraisemblablement que dans le compartiment dans lequel ils ont été produits. Un point important est que la rénine active, si elle diffuse librement dans le plasma, s’adsorbe dans les tissus, en particulier dans la paroi artérielle. Dans le compartiment interstitiel, les résidus mannose-6 phosphate retiennent la rénine par des liaisons électrostatiques de faible affinité [2], cette liaison conduisant à un enrichissement en rénine et en angiotensine du milieu interstitiel par rapport au plasma [3]. Un récepteur membranaire ayant une forte affinité pour la prorénine et la rénine active [4] a par ailleurs récemment été mis en évidence. Son implication physiologique et physiopathologique doit encore être précisée.
On peut donc différencier l’interaction rénine-angiotensinogène dans le plasma, à l’origine de la production d’angiotensine I, rapidement convertie en angiotensine II par l’enzyme de conversion abondamment présente dans le plasma et l’endothélium, et l’interaction péricellulaire rénine-angiotensinogène, tous deux adsorbés dans les tissus où l’expression du récepteur de la rénine et l’expression non constitutive de l’enzyme de conversion sont limitantes de la production d’angiotensine II [5] (Figure 1).
Figure 1
Compartimentation du système rénine-angiotensine.
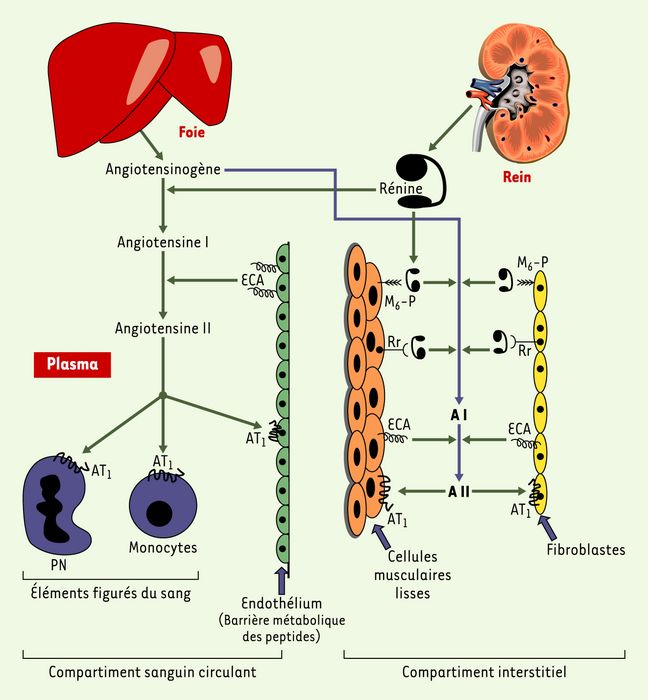
Les protéines sécrétées, rénine et angiotensinogène, diffusent dans les deux compartiments plasmatique et interstitiel, alors que les peptides, angiotensines I et II, sont métabolisés dans le compartiment où ils ont été produits. M6-P : récepteur de faible affinité du mannose 6-phosphate ; Rr : récepteur de forte affinité de la rénine ; ECA : enzyme de conversion de l’angiotensine ; AT1 : récepteur de type 1 de l’angiotensine II ; PN : polynucléaire.
Signalisation induite par l’angiotensine II dans les cellules cibles
L’angiotensine II agit sur ses cellules cibles par l’intermédiaire de récepteurs à sept domaines transmembranaires dont il existe deux types, AT1 et AT2. Ce dernier est surtout impliqué dans l’embryogenèse, et son rôle dans le remodelage vasculaire ne sera pas abordé ici (pour revue, voir [6]). Le récepteur AT1 est présent sur les monocytes, les polymorphonucléaires, les cellules endothéliales, les cellules musculaires lisses et les fibroblastes. Dans ces cellules, le récepteur AT1 est couplé aux phospholipases C, D et A2 (Figure 2).
Figure 2
Signalisation intracellulaire par l’angiotensine II.
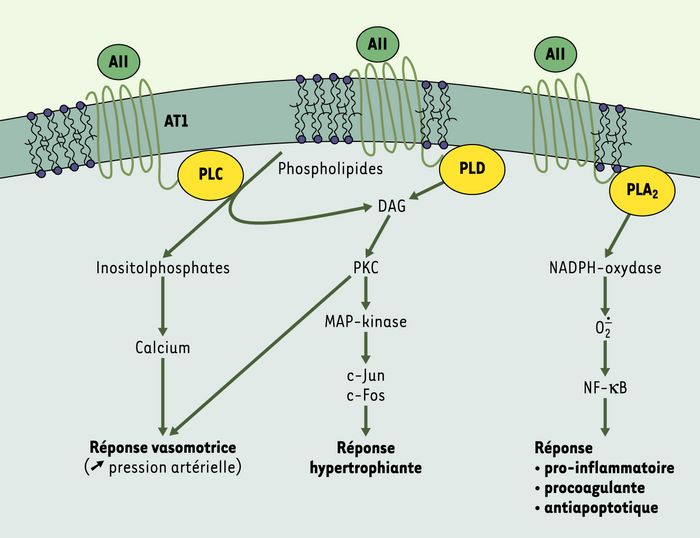
AII : angiotensine II ; AT1 : récepteur de type 1 de l’angiotensine II ; PL : phospholipases ; DAG : diacylglycérol ; PK : protéine kinase ; MAP-kinases : mitogen-activated protein-kinases ; NF-κB : nuclear factor κB.
Schématiquement, on peut d’abord identifier la signalisation fonctionnelle immédiate, en réponse à l’interaction entre l’angiotensine II et son récepteur AT1, par l’intermédiaire de l’activation de la phospholipase C, de la libération de phospho-inositides, de la mobilisation du calcium et, pour partie, de l’activation de la protéine kinase C. C’est cette réponse fonctionnelle qui est responsable des effets vasomoteurs de l’angiotensine II, via la cellule musculaire lisse artérielle, et de la libération immédiate de monoxyde d’azote par l’endothélium [7].
Dans un second temps, la protéine kinase C, activée par le diacylglycérol (issu de la déphosphorylation de l’acide phosphatidique produit par la phospholipase D), agit sur la régulation de l’expression génique, via le complexe AP-1, de c-Jun, de c-Fos et des MAP-kinases (mitogen-activated protein-kinases) [8]. Cette activation aboutit à une augmentation globale de la synthèse protéique, en particulier des facteurs de croissance FGF (fibroblast growth factor) et TGF-β (transforming growth factor-β) [9], de protéines nucléaires, de protéines de la matrice extracellulaire et de protéines sécrétées comme les antiprotéases (PAI-1, plasminogen-activator inhibitor). Ce sont ces mécanismes qui sont impliqués dans les effets hypertrophiants de l’angiotensine II et dans la facilitation d’entrée en mitose qu’elle induit.
Le troisième niveau d’activation emprunte la voie de la phospholipase A2, qui s’accompagne de la libération d’eicosanoïdes et de la production d’un stress oxydatif dans les cellules cibles. La voie des eicosanoïdes (impliquant un substrat, l’acide arachidonique, et des enzymes, cyclooxygénases et lipoxygénases, prostaglandines - et thromboxanes - synthétases) aboutit à la production de prostaglandines qui ont un un effet vasodilatateur et antiagrégant. Cette voie assure aussi la synthèse du thromboxane A2, de leucotriènes ou du PAF (platelet-activating factor)-acéther, facteurs vasoconstricteurs et proagrégants, capables de surcroît d’augmenter la perméabilité endothéliale.
L’augmentation du stress oxydatif induit par l’angiotensine II dans ses cellules cibles est également principalement liée à l’activation de la phospholipase A2 [10], associée à la NADPH oxydase membranaire productrice d’anions superoxydes [11]. Une interaction avec les éléments mitochondriaux producteurs de formes réactives de l’oxygène n’est par ailleurs pas exclue. L’augmentation du stress oxydatif dans la cellule provoque l’activation du système NF (nuclear factor)-κ-B qui, transloqué au noyau, induit la transcription de gènes codant pour des facteurs pro-inflammatoires (IL [interleukine]-6, VCAM [vascular cell-adhesion molecule]…) [12] et procoagulants (facteur tissulaire) [13].
Dans le contexte du remodelage vasculaire, les cibles de l’angiotensine II sont les leucocytes circulants et les cellules endothéliales (pour l’angiotensine II plasmatique), et les cellules musculaires lisses et les fibroblastes adventitiels de la paroi artérielle (pour l’angiotensine II interstitielle). Si les cascades biochimiques de signalisation sont communes à ces différents types cellulaires, la réponse finale dépend de la physiologie propre à chaque type cellulaire : augmentation de la perméabilité et synthèse de molécules d’adhérence pour les cellules endothéliales [14], hypertrophie et sécrétion de cytokines pro-inflammatoires pour les cellules musculaires lisses, production de TGF-β et de collagène pour les fibroblastes, activation des leucocytes.
Toutes ces données, essentiellement obtenues in vitro, ne tiennent généralement pas compte des contre-régulations physiologiques existant in vivo (en particulier de celle assurée par le monoxyde d’azote endothélial, produit in vivo en réponse au cisaillement endothélial), ni des modifications phénotypiques des cellules (phénotype sécrétoire pour les cellules musculaires lisses, par exemple). Toutefois, elle sont probablement représentatives d’un certain nombre de phénomènes chroniques observés in vivo, en pathologie, lorsque les cellules vasculaires sont activées par les conditions environnementales.
Système rénine-angiotensine et remodelage vasculaire
L’activation du système rénine-angiotensine in vivo peut être étudiée de deux façons : soit en analysant les effets de l’activation du système sur la structure vasculaire (dans des cas d’hypertension rénovasculaire expérimentale ou chez des animaux transgéniques surexprimant la rénine), soit en étudiant les conséquences de l’inhibition de l’angiotensine II dans des modèles ne dépendant pas directement de l’activation du système rénine-angiotensine. Dans tous les cas, il est toujours difficile de séparer complètement la réponse fonctionnelle sur la pression artérielle de la réponse structurale sur la paroi des vaisseaux.
Classiquement, la production d’angiotensine II conduit à une hypertension artérielle et à une hypertrophie de la paroi artérielle. Cette dernière s’accompagne d’une augmentation de la synthèse de matrice extracellulaire [15] et de la sécrétion plasmatique d’antiprotéases telles que PAI-1 et TIMP (tissue inhibitor of metalloproteinases) [16]. L’hypertension, en augmentant la « tenségrité » (tension interne réglée par la conformation du cytosquelette) des cellules musculaires lisses liée à leur adhérence à la matrice extracellulaire, s’ajoute ici aux effets directs de l’angiotensine II sur la trophicité des cellules vasculaires. Inversement, lorsque l’hypertension artérielle n’est pas directement liée à l’activation du système rénine-angiotensine, comme chez le rat spontanément hypertendu ou dans le modèle d’intoxication à la nitroarginine (suppression de la production endothéliale de monoxyde d’azote, NO, au potentiel vasodilatateur), le blocage du système rénine-angiotensine entraîne une baisse importante de la pression artérielle et de la trophicité de la paroi artérielle [17].
Angiotensine II et fibrose
Au-delà de l’hypertrophie vasculaire, l’angiotensine II est un facteur profibrosant capable d’activer directement, comme indirectement, les fibroblastes interstitiels [18]. Directement, l’angiotensine II augmente l’activité de synthèse de matrice extracellulaire par les fibroblastes en culture tandis que, indirectement, elle facilite l’accumulation périvasculaire de cellules inflammatoires productrices de TGF-β limitant le phénomène inflammatoire et activant la fibrose.
Les interactions entre système rénine-angiotensine et complications vasculaires du diabète constituent un autre exemple de l’implication du TGF-β. Les inhibiteurs de l’enzyme de conversion et les antagonistes de l’angiotensine II [19, 20] ont un effet protecteur vis-à-vis des complications vasculaires du diabète, entre autres via une diminution de l’expression du TGF-β. De même, le déterminisme génétique d’expression de l’enzyme de conversion est un des facteurs reconnus de susceptibilité aux complications vasculaires du diabète [21]. Le diabète et l’angiotensine II ont des effets additifs sur l’expression du TGF-β, ce dernier constituant un médiateur de choix entre ces deux facteurs et le remodelage vasculaire pathologique. Nous avons récemment montré une relation entre l’expression tissulaire de l’enzyme de conversion génétiquement déterminée et induite par le diabète et celle du TGF-β dans les artérioles rénales [22].
Un autre exemple de la relation entre l’activation du système rénine-angiotensine et la fibrose cardiovasculaire est celui des valves cardiaques et de la cicatrice fibreuse postinfarctus. En physiologie, les myofibroblastes des valves, activés par la contrainte mécanique de chaque battement cardiaque, expriment fortement l’enzyme de conversion [23]. Il a récemment été montré que cette expression est augmentée dans les valves pathologiques, en association avec les dépôts de LDL (low density lipoprotein)-apo B [24], ainsi que dans les lésions d’athérosclérose ; dans le plasma, l’enzyme de conversion est également associée avec la fraction LDL. Dans le même ordre d’idées, la cicatrice fibreuse post-infarctus, hémodynamiquement contrainte, exprime l’enzyme de conversion [25, 26], et les antagonistes de l’angiotensine II diminuent l’expression de TGF-β dans la cicatrice [27].
De façon analogue, les inhibiteurs du système rénine-angiotensine diminuent la prolifération intimale des cellules musculaires lisses secondaire à une angioplastie. Par ailleurs, le polymorphisme d’expression de l’enzyme de conversion influence le taux de resténose après angioplastie chez l’animal [28] comme chez l’homme [29]. Ces effets sont essentiellement dus à l’angiotensine II interstitielle, produite localement, au niveau des cellules musculaires lisses ou des fibroblastes, à partir de la rénine et de l’angiotensinogène adsorbés dans les tissus. Le rôle dans ce contexte du récepteur de la rénine, récemment mis en évidence, reste à préciser.
L’angiotensine II du compartiment plasmatique interagit avec les cellules endothéliales et les leucocytes circulants. L’interaction entre l’angiotensine II et l’endothélium peut induire une première réponse fonctionnelle de libération de monoxyde d’azote via les récepteurs AT1 [7], diminuant transitoirement la pression artérielle. Les principaux effets chroniques de l’interaction de l’angiotensine II sur l’endothélium, en particulier dans des contextes de faible production de NO (endothélium peu stimulé par le cisaillement hémodynamique, déficit d’activité de la NO synthétase en rapport avec l’athérome), sont assurés par l’activation de la phospholipase A2 et du stress oxydatif intracellulaire. L’angiotensine II induit alors la production d’eicosanoïdes et un phénotype pro-inflammatoire de la cellule endothéliale.
Angiotensine II, inflammation et athérosclérose
L’angiotensine II circulante peut interférer avec les éléments figurés du sang, en particulier avec les monocytes-macrophages. L’angiotensine II active les monocytes, mais également les polymorphonucléaires neutrophiles, via leur récepteurs AT-1 [30]. L’angiotensine II induit une dégranulation partielle des polymorphonucléaires neutrophiles, ainsi que la production d’ions superoxydes et la sécrétion de leucotriènes.
Un exemple de cet effet sur les leucocytes a été apporté par une étude menée chez des souris invalidées pour le gène de l’apolipoprotéine E (Apo E-/-), donc susceptibles à l’athérosclérose : la perfusion directe d’angiotensine II n’induit qu’une augmentation très limitée de la pression artérielle, mais provoque la formation d’anévrismes de l’aorte [31]. Ces derniers sont associés à une disparition des cellules musculaires lisses artérielles, à une destruction de la matrice extracellulaire et à une infiltration importante de cellules inflammatoires productrices de protéases (activateurs du plasminogène, métalloprotéinases) [32]. Si ces résultats semblent paradoxaux lorsqu’on connaît les conséquences de l’activation du système rénine-angiotensine interstitiel sur l’hypertrophie de la paroi artérielle et sur l’augmentation de la sécrétion d’antiprotéases par les cellules musculaires lisses, ils s’expliquent par un effet préférentiel de l’angiotensine II circulante sur les leucocytes. Une greffe de moelle osseuse provenant de souris invalidées pour le gène codant pour le récepteur AT1 de l’angiotensine II (souris AT1-/-) prévient la formation de ces anévrismes [33].
À l’inverse, les inhibiteurs du système rénine-angiotensine freinent le développement de l’athérosclérose chez les souris apo E-/- et les lapins hypercholestérolémiques. Il a été montré que l’irbesartan, un antagoniste du récepteur AT1 de l’angiotensine II, diminue les lésions d’athérosclérose chez la souris Apo E-/- en diminuant le phénotype inflammatoire de la plaque [34].
Conclusions
Le mode d’action du système rénine-angiotensine sur le remodelage vasculaire est complexe d’un point de vue biologique ; il dépend de plusieurs facteurs, parmi lesquels les plus importants sont probablement la nature du compartiment dans lequel l’angiotensine Il est produite et l’état de réceptivité des cellules cibles. Cette action sur le remodelage est fonction de la capacité du tissu pathologique à fixer la rénine et à exprimer l’enzyme de conversion in situ.
Les inhibiteurs du système rénine-angiotensine sont apparus il y a plus de vingt ans avec les inhibiteurs de l’enzyme de conversion de l’angiotensine ; les antagonistes de l’angiotensine II sont, quant à eux, plus récents. Au-delà de leurs effets fonctionnels immédiats sur la pression artérielle, les inhibiteurs du système, qui ont une biodisponibilité satisfaisante vis-à-vis de leurs cibles circulantes et tissulaires, sont efficaces pour prévenir les effets de l’angiotensine II sur le remodelage vasculaire. Cependant, aucune de ces deux classes moléculaires n’est complètement spécifique, ni complètement inhibitrice aux doses employées en clinique. Leur association a un effet additif sur la qualité d’inhibition du système en agissant à des niveaux différents de la cascade biochimique du système [35]. Il est également probable que la biodisponibilité des molécules de ces deux classes, dans le compartiment interstitiel et le compartiment circulant, sont assez différentes. Dans ce contexte, s’il est intéressant de tester leur additivité sur l’hypertension artérielle, ce le sera encore plus sur le remodelage vasculaire.
Parties annexes
Références
- 1. Goldblatt H. The renal origin of hypertension. Physiol Rev 1947 ; 27 : 120-65.
- 2. van Kesteren CA, Danser AH, Derkx FH, et al. Mannose 6-phosphate receptor-mediated internalization and activation of prorenin by cardiac cells. Hypertension 1997 ; 30 : 1389-96.
- 3. de Lannoy LM, Danser AH, van Kats JP, et al. Renin-angiotensin system components in the interstitial fluid of the isolated perfused rat heart. Local production of angiotensin I. Hypertension 1997 ; 29 : 1240-51.
- 4. Nguyen G, Delarue F, Burckle C, et al. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J Clin Invest 2002 ; 109 : 1417-27.
- 5. Coulet F, Gonzalez W, Boixel C, et al. Endothelium-independent conversion of angiotensin I by vascular smooth muscle cells. Cell Tissue Res 2001 ; 303 : 227-34.
- 6. Levy BI. Can angiotensin II type 2 receptors have deleterious effects in cardiovascular disease? Implications for therapeutic blockade of the renin-angiotensin system. Circulation 2004 ; 109 : 8-13.
- 7. Pueyo ME, N’Diaye N, Michel JB. Angiotensin II-elicited signal transduction via AT1 receptors in endothelial cells. Br J Pharmacol 1996 ; 118 : 79-84.
- 8. Berk BC, Corson MA. Angiotensin II signal transduction in vascular smooth muscle : role of tyrosine kinases. Circ Res 1997 ; 80 : 607-16.
- 9. Weigert C, Brodbeck K, Klopfer K, et al. Angiotensin II induces human TGF-beta 1 promoter activation : similarity to hyperglycaemia. Diabetologia 2002 ; 45 : 890-8.
- 10. Griendling KK, Ushio-Fukai M. Reactive oxygen species as mediators of angiotensin II signaling. Regul Pept 2000 ; 91 : 21-7.
- 11. Pagano PJ, Chanock SJ, Siwik DA, et al. Angiotensin II induces p67phox mRNA expression and NADPH oxidase superoxide generation in rabbit aortic adventitial fibroblasts. Hypertension 1998 ; 32 : 331-7.
- 12. Kranzhofer R, Schmidt J, Pfeiffer CA, et al. Angiotensin induces inflammatory activation of human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 1999 ; 19 : 1623-9.
- 13. Brown NJ, Vaughan DE. Prothrombotic effects of angiotensin. Adv Intern Med 2000 ; 45 : 419-29.
- 14. Pueyo ME, Gonzalez W, Nicoletti A, et al. Angiotensin II stimulates endothelial vascular cell adhesion molecule-1 via nuclear factor-kappaB activation induced by intracellular oxidative stress. Arterioscler Thromb Vasc Biol 2000 ; 20 : 645-51.
- 15. Brilla CG, Zhou G, Matsubara L, Weber KT. Collagen metabolism in cultured adult rat cardiac fibroblasts : response to angiotensin II and aldosterone. J Mol Cell Cardiol 1994 ; 26 : 809-20.
- 16. Brown NJ, Kumar S, Painter CA, Vaughan DE. ACE inhibition versus angiotensin type 1 receptor antagonism : differential effects on PAI-1 over time. Hypertension 2002 ; 40 : 859-65.
- 17. Henrion D, Dowell FJ, Levy BI, Michel JB. In vitro alteration of aortic vascular reactivity in hypertension induced by chronic NG-nitro-L-arginine methyl ester. Hypertension 1996 ; 28 : 361-6.
- 18. Pagano PJ, Clark JK, Cifuentes-Pagano ME, et al. Localization of a constitutively active, phagocyte-like NADPH oxidase in rabbit aortic adventitia : enhancement by angiotensin II. Proc Natl Acad Sci USA 1997 ; 94 : 14483-8.
- 19. Lewis EJ, Hunsicker LG, Clarke WR, et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 2001 ; 345 : 851-60.
- 20. Parving HH, Lehnert H, Brochner-Mortensen J, et al. The effect of irbesartan on the development of diabetic nephropathy in patients with type 2 diabetes. N Engl J Med 2001 ; 345 : 870-8.
- 21. Marre M, Jeunemaitre X, Gallois Y, et al. Contribution of genetic polymorphism in the renin-angiotensin system to the development of renal complications in insulin-dependent diabetes : génétique de la néphropathie diabétique (GENEDIAB) study group. J Clin Invest 1997 ; 99 : 1585-95.
- 22. Pueyo ME, Challah, M, Gaugier, D, et al. TGF-?1 production is correlated with genetically determined angiotensin-converting enzyme expression in congenic rats. Diabetes 2004 (sous presse).
- 23. Yamada H, Fabris B, Allen AM, et al. Localization of angiotensin converting enzyme in rat heart. Circ Res 1991 ; 68 : 141-9.
- 24. Brien KD, Shavelle DM, Caulfield MT, et al. Association of angiotensin-converting enzyme with low-density lipoprotein in aortic valvular lesions and in human plasma. Circulation 2002 ; 106 : 2224-30.
- 25. Johnston CI, Mooser V, Sun Y, Fabris B. Changes in cardiac angiotensin converting enzyme after myocardial infarction and hypertrophy in rats. Clin Exp Pharmacol Physiol 1991 ; 18 : 107-10.
- 26. Gaertner R, Prunier F, Philippe M, et al. Scar and pulmonary expression and shedding of ACE in rat myocardial infarction. Am J Physiol Heart Circ Physiol 2002 ; 283 : H156-64.
- 27. Sun Y, Zhang JQ, Zhang J, Ramires FJ. Angiotensin II, transforming growth factor-beta1 and repair in the infarcted heart. J Mol Cell Cardiol 1998 ; 30 : 1559-69.
- 28. Challah M, Villard E, Philippe M, et al. Angiotensin I-converting enzyme genotype influences arterial response to injury in normotensive rats. Arterioscler Thromb Vasc Biol 1998 ; 18 : 235-43.
- 29. Amant C, Bauters C, Bodart JC, et al. Dallele of the angiotensin I-converting enzyme is a major risk factor for restenosis after coronary stenting. Circulation 1997 ; 96 : 56-60.
- 30. Paragh G, Szabo J, Kovacs E, et al. Altered signal pathway in angiotensin II-stimulated neutrophils of patients with hypercholesterolaemia. Cell Signal 2002 ; 14 : 787-92.
- 31. Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest 2000 ; 105 : 1605-12.
- 32. Wang YX, Martin-McNulty B, Freay AD, et al. Angiotensin II increases urokinase-type plasminogen activator expression and induces aneurysm in the abdominal aorta of apolipoprotein E-deficient mice. Am J Pathol 2001 ; 159 : 1455-64.
- 33. Manning MW, Cassi LA, Huang J, et al A. Abdominal aortic aneurysms : fresh insights from a novel animal model of the disease. Vasc Med 2002 ; 7 : 45-54.
- 34. Dol F, Martin G, Staels B, et al. Angiotensin AT1 receptor antagonist irbesartan decreases lesion size, chemokine expression, and macrophage accumulation in apolipoprotein E-deficient mice. J Cardiovasc Pharmacol 2001 ; 38 : 395-405.
- 35. Menard J, Campbell DJ, Azizi M, Gonzales MF. Synergistic effects of ACE inhibition and Ang II antagonism on blood pressure, cardiac weight, and renin in spontaneously hypertensive rats. Circulation 1997 ; 96 : 3072-8.
Liste des figures
Figure 1
Compartimentation du système rénine-angiotensine.
Les protéines sécrétées, rénine et angiotensinogène, diffusent dans les deux compartiments plasmatique et interstitiel, alors que les peptides, angiotensines I et II, sont métabolisés dans le compartiment où ils ont été produits. M6-P : récepteur de faible affinité du mannose 6-phosphate ; Rr : récepteur de forte affinité de la rénine ; ECA : enzyme de conversion de l’angiotensine ; AT1 : récepteur de type 1 de l’angiotensine II ; PN : polynucléaire.
Figure 2
Signalisation intracellulaire par l’angiotensine II.
AII : angiotensine II ; AT1 : récepteur de type 1 de l’angiotensine II ; PL : phospholipases ; DAG : diacylglycérol ; PK : protéine kinase ; MAP-kinases : mitogen-activated protein-kinases ; NF-κB : nuclear factor κB.