Corps de l’article
Parmi les avances staturales congénitales, deux syndromes cliniquement voisins avaient été identifiés et considérés comme deux entités distinctes: le syndrome de Sotos, et le syndrome de Weaver.
En 2002, la découverte de l’implication du gène NSD1 dans le syndrome de Sotos ((→) m/s 2002, n°8-9, p.825), avec la mise en évidence de mutations dans plus 70 % des cas japonais [1] a été suivie par des recherches analogues dans d’autres pays. Deux études européennes viennent d’être publiées, l’une au Royaume-Uni, l’autre en France, et font le point sur la fréquence et le type des mutations du gène NSD1 dans les macrosomies congénitales [2, 3]. Elles révèlent une même origine génétique pour les deux syndromes, Sotos et Weaver, du moins dans un certain nombre de cas, permettant de considérer comme alléliques ces formes syndromiques d’avance staturale.
Le syndrome de Sotos
Décrit en 1964, le syndrome de Sotos (SS) ou gigantisme cérébral se caractérise par une macrosomie pré- et post-natale, avec avance de l’âge osseux. L’aspect du visage (macrocéphalie avec front bombé et implantation haute des cheveux qui sont peu fournis, menton proéminent) est caractéristique. Toutefois, le diagnostic n’est pas toujours évident, d’autant que les courbes de croissance se normalisent avec l’âge, surtout chez les filles dont la puberté est parfois précoce. Des signes complémentaires sont rapportés ainsi qu’une prédisposition à des cancers (tératome sacro-coccygien, neuroblastome, cancer gastrique).
Bien que la plupart des cas soient sporadiques, quelques familles avec transmission autosomique dominante ont été décrites. Mais la mise en évidence d’un locus n’a pu se faire que grâce à une translocation réciproque équilibrée de novo survenue chez une petite japonaise de 15 mois [4]. Un des points de cassure, en 5q35, a permis de trouver une microdélétion avec perte du gène NSD1. Celui-ci avait été identifié auparavant en raison d’une translocation (5;11)(q35;p15) survenant de façon non aléatoire dans des leucémies myéloïdes aiguës de l’enfant (créant une protéine de fusion associant NUP98, une nucléoporine, avec le produit de NSD1).
Le gène NSD1
Ce gène (dont la formule développée est la suivante: nuclear receptor-binding Su-var, enhancer of zeste, and trithorax domain protein 1) code pour un des co-régulateurs du récepteur des androgènes, aussi appelé ARA267 (androgen receptor associated coregulator).
ARA267 interagit avec ARA24 et PCAF (p300/CBP-associated factor) entre autres, pour moduler la transcription. Sa fonction n’est pas complètement élucidée, mais il apparaît comme un intermédiaire transcriptionnel capable d’influencer positivement ou négativement la transcription selon le contexte cellulaire. Il code pour une protéine de 2596 acides aminés. Celle-ci a 85% d’identité de séquence avec la protéine murine et contient plusieurs domaines fonctionnels: un domaine SET (SU[VAR]3-9,E[Z]), trithorax, initialement identifié dans des gènes de drosophile impliqués dans une régulation dépendant de la chromatine au cours du développement. À côté se trouve un domaine SAC (SET-associated Cys-rich). Or, la combinaison de ces deux domaines se rencontre dans des protéines ayant une fonction d’histone-méthyl transférase (HMTases). Donc la protéine NSD1 agit peut-être en modifiant les histones, la régulation et la maintenance des états de la chromatine. Il existe aussi cinq homéodomaines PHD (plant homeodomain, motif analogue aux doigts de zinc) qui existent, là encore, dans des protéines agissant au niveau chromatinien. Les domaines PHD ont un motif consensus C4HC3 que l’on retrouve ici dans les premiers PHD. Le cinquième homéodomaine contient une histidine au lieu d’une cystéine et doit être appelé PHD-H2. Enfin, il existe deux domaines PWWP (proline-tryptophane-tryptopha-ne-proline) que l’on trouve dans des facteurs régulateurs et qui interviennent dans les interactions protéine-protéine (Figure 1).
Le syndrome de Weaver
Le syndrome de Weaver (WS), décrit un peu plus tard, est une entité clinique distincte du syndrome de Sotos. En effet, même s’il se caractérise par les mêmes signes que le syndrome de Sotos, il en diffère par la dysmorphie faciale (micrognathie et long filtrum), une hyperlaxité avec excès de peau, modifiant l’aspect du visage et s’accompagnant d’une voix rauque. Il existe aussi une camptodactylie[1], des ongles hypoplasiques, et une dysplasie dentaire. Toutefois, l’individualité du syndrome de Weaver fut parfois mise en doute, en particulier par Opitz [5].
La rareté des syndromes de Weaver et leur caractère sporadique ne permettait pas de trouver un locus. Il fallut donc attendre l’isolement d’un gène dans le syndrome de Sotos pour pouvoir conclure.
Macrosomies et NSD1
Les deux études européennes récentes portent sur 114 cas de macrosomie (Tableau I). Ils se répartissent en quatre groupes: (1) 60 cas de syndrome de Sotos typiques; (2) 23 cas évocateurs mais ne présentant pas tous les signes; (3) 13 cas de syndrome de Weaver; (4) 18cas de macrosomie ne correspondant à aucun autre syndrome connu, sauf un cas de syndrome de Marshall-Smith. Les résultats montrent que le gène NSD1 est impliqué dans environ 70 % des cas de syndrome de Sotos. Ce pourcentage est équivalent à celui de la première étude japonaise (77% des cas de Sotos typiques) mais, alors que ceux-ci avaient presque tous une grande délétion de 2,2Mb, dans les deux études européennes, il s’agit surtout de mutations tronquantes. Neuf délétions seulement ont été observées, plus petites que la délétion japonaise. L’étude moléculaire des parents, lorsqu’elle a été possible, montre que les délétions et les mutations sont survenues de novo. Dans l’étude française, le chromosome porteur de la délétion était surtout d’origine paternelle (avec augmentation de l’âge du père à la naissance). Dans l’étude britannique, il est très probable que les mutations faux-sens, situées dans la moitié carboxy-terminale du gène, entre les exons 13 et 23 sont pathogènes puisqu’elles sont apparues de novo (l’étude chez les parents fut réalisée dans sept des neuf cas). Quant aux mutations ponctuelles dans les WS, elles sont situées dans des régions carboxy-terminales (Figure 1).
Tableau I
Nombre de malades, fréquence et types de mutations observées dans les deux études européennes.
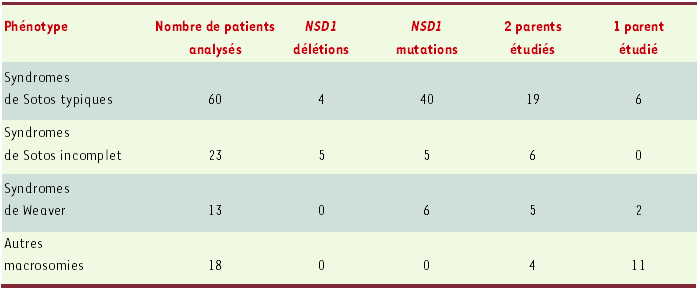
Figure 1
Représentation schématique du gène NSD1 avec les mutations observées dans l’étude britannique.

Les différents domaines sont mentionnés et colorés. Les mutations des syndromes de Sotos sont en rouge et celles des syndomes de Weaver en bleu.
Discussion
L’existence de délétions indique que les syndromes de Sotos et de Weaver sont la conséquence d’une haploinsuffisance. Chez les malades de type Sotos porteurs d’une délétion, le retard mental est nettement plus sévère, sans aucune acquisition du langage. La dysmorphie faciale semble plus marquée; en revanche, l’avance staturale est variable et ne semble pas un critère majeur. La présence fréquente d’une délétion au Japon ne peut s’expliquer par un effet fondateur puisqu’il s’agit d’accidents survenus de novo. Six des treize patients Weaver étudiés étaient porteurs de mutations ponctuelles situées dans la région carboxy-terminale. Il n’existe aucune différence clinique entre les cas de WS avec mutations du gène NSD1 et les cas où le gène semble intact. Il reste désormais à comprendre par quel mécanisme la perte d’un régulateur transcriptionnel bifonctionnel peut entraîner une macrosomie et les anomalies qui lui sont associées, et comment il peut aboutir à deux syndromes qui, malgré leurs nombreux points communs, représentent cependant des entités cliniques distinctes.
Enfin, bien que NSD1 soit certainement le gène majeur dans ces deux maladies, des recherches doivent être poursuivies dans les cas où NSD1 ne semble pas impliqué.
Parties annexes
Note
-
[1]
Flexion permanente d’un ou plusieurs doigts de la main.
Références
- 1. Kurotaki N, Imaizumi K, Harada N, et al. Haplo insufficiency of NSD1 causes Sotos syndrome. Nature 2002 ; 30 : 305-6.
- 2. Douglas J, Hanks S, Temple K, et al. NSD1 mutations are the major cause of Sotos syndrome and occur in some cases of Weaver syndrome but are rare in other overgroth phenotypes. Am J Hum Genet 2003 ; 27 : 132-43.
- 3. Rio M, Clech L, Amiel J, et al. Spectrum of NSD1 mutations in Sotos and Weaver syndromes. J Med Genet 2003 ; 40 : 436-40.
- 4. Imaizumi K, Kimura J, Matsuo M, et al. Sotos syndrome associated with a de novo balanced reciprocal translocation t(5 ;8)(q35 ;q24.1). Am J Hum Genet 2002 ; 107 : 58-60.
- 5. Opitz JM, Weaver DW, Reynolds JF. The syndrome of Sotos and Weaver : reports and review. Am J Med Genet 1998 ; 79 : 294-304.
Liste des figures
Figure 1
Représentation schématique du gène NSD1 avec les mutations observées dans l’étude britannique.
Les différents domaines sont mentionnés et colorés. Les mutations des syndromes de Sotos sont en rouge et celles des syndomes de Weaver en bleu.
Liste des tableaux
Tableau I
Nombre de malades, fréquence et types de mutations observées dans les deux études européennes.