Résumés
Résumé
Les accidents ischémiques et les crises épileptiques font partie des causes majeures de mort neuronale. Malgré les nombreuses recherches sur la neuroprotection et la découverte de nouvelles molécules capables de bloquer certains des événements délétères chez l’animal, peu de traitements sont disponibles pour lutter contre la mort neuronale induite par ces maladies. Le plus efficace reste le traitement thrombolytique en phase aiguë des accidents ischémiques cérébraux, qui ne peut toutefois être administré que chez une très faible population de patients. La prévention visant à réduire l’incidence des facteurs de risque reste donc la principale stratégie thérapeutique. La tolérance cérébrale est un phénomène endogène reposant sur l’induction préventive de la résistance neuronale. La compréhension de ses mécanismes permettrait d’identifier de nouvelles cibles thérapeutiques. Les inducteurs pharmacologiques de la tolérance cérébrale devraient conduire à la même neuroprotection et à la mise en oeuvre de nouvelles thérapeutiques.
Summary
Ischemia and seizures are common diseases that result in neuronal death. To-date, there are no available treatments to block or reverse neuronal death pathways in patients who suffer from these diseases. All drugs that have been shown to be neuroprotective in animal models have failed in human trials. Therefore, the potential of preventative strategies for therapy is increasingly explored. Experimental studies have demonstrated that a brief cerebral ischemic insult, that is not harmful by itself, results in a temporary protective adaptation in the brain against a subsequent ischemic episode that would otherwise be lethal. This process, termed ischemic preconditioning, has been confirmed in different models of cerebral ischemia. A similar phenomenon observed after a mild epileptic insult conferred a transitory tolerance to a subsequent epileptic episode. This process is termed epileptic tolerance. Other stresses, like hyperthermia or spreading depression, also enhanced brain resistance to detrimental effects of ischemic or epileptic injury. Recently, a cross tolerance between ischemia and epilepsy has been reported. Also, some retrospective studies in humans suggest that endogenous ischemic preconditioning exists in the brain. Altogether these insights of brain tolerance point to the future discovery of potentially useful targets for acute neuroprotection as well as preventive therapy.
Corps de l’article
Les premières définitions du préconditionnement et de la tolérance ischémiques ont été établies au niveau cardiaque par C.E. Murry et al. en 1986 [1] ((→) m/s 2004, n° 5, p. 521).
Depuis, ce phénomène de protection endogène a été mis en évidence chez la plupart des mammifères, y compris chez l’homme où il est actuellement utilisé en chirurgie cardiaque. Parallèlement, la notion de tolérance à l’ischémie s’est étendue à d’autres organes, et en particulier au cerveau. Dès 1986, A. Schurr et al. ont montré in vitro qu’une courte période d’anoxie protège l’hippocampe d’une anoxie plus longue, normalement délétère pour les neurones. Ce processus d’adaptation des neurones présentait les caractéristiques qui servent aujourd’hui à définir le préconditionnement [2]. Mais il a fallu attendre 1990 pour que le terme de tolérance cérébrale ischémique soit utilisé, et cinq ans de plus pour que la tolérance épileptique soit définie par analogie avec la tolérance ischémique : c’est-à-dire qu’une crise épileptique modérée protège le cerveau d’une crise normalement délétère pour les neurones [3, 4].
Dans les premières expériences visant à établir un état de tolérance contre un stimulus pathogène, l’inducteur était de même type que le stimulus létal, d’où le terme de préconditionnement. Seule l’intensité des stimulus variait, le stimulus inducteur devant être infraliminaire au seuil du dommage irréversible et le second stimulus étant supposé létal pour les neurones. Les imprécisions dans la terminologie ont engendré des confusions entre la tolérance - qui correspond à la fenêtre de protection contre une maladie - et le préconditionnement, qui est l’inducteur de celle-ci. Cela a compliqué la compréhension du phénomène et masqué son important potentiel thérapeutique. L’avènement de nouvelles technologies et une meilleure approche des exemples historiques de la tolérance cérébrale ischémique et épileptique devraient faciliter les futures investigations.
La tolérance ischémique cérébrale
Le phénomène de tolérance ischémique est d’abord un concept expérimental, décrit principalement dans le coeur et le cerveau, mais aussi dans de nombreux autres organes. L’induction et le maintien de la tolérance impliquent des effecteurs moléculaires dont la plupart semblent communs aux différents préconditionnements. Cependant, les spécificités liées à la régulation de l’excitabilité neuronale font de la tolérance cérébrale un phénomène unique.
Les premières études in vivo ont montré qu’une ischémie cérébrale modérée déclenche un mécanisme de protection endogène : le préconditionnement cérébral de type ischémique réduit la neurodégénérescence normalement induite par une ischémie cérébrale sévère lorsque le cerveau n’est pas préconditionné [3] (Figures 1 et 2).
Figure 1
Critères définissant les concepts du préconditionnement et de la tolérance cérébrale.

Il existe plusieurs types de préconditionnement cérébral, définis en fonction de leur inducteur (la première agression cérébrale, qui doit dans tous les cas être modérée). Il existe toujours un délai, impliquant des changements biochimiques, avant que la fenêtre de protection ne s’établisse. Les deux fenêtres de protection généralement décrites sont transitoires et ont des profils temporels distincts : la fenêtre précoce est de courte durée et apparaît rapidement après le préconditionnement ; la fenêtre retardée est quant à elle effective entre 24 et 96 heures après le préconditionnement.
Figure 2
Protection des neurones de l’hippocampe par différents types de préconditionnement et analogie entre les tolérances ischémique et épileptique.

La tolérance cérébrale ischémique décrite dans cette figure est induite par un préconditionnement ischémique (ischémie cérébrale modérée). Ce dernier réduit la neurodégénérescence au niveau des couches du CA1 de l’hippocampe induite par une ischémie cérébrale sévère lorsque le cerveau n’est pas préconditionné. De même, la tolérance épileptique est induite par un préconditionnement épileptique (crise épileptique modérée) qui préserve les neurones des couches du CA3 de l’hippocampe détruits lors d’une crise épileptique sévère. Le préconditionnement pharmacologique, par les activateurs des canaux KATP ou des récepteurs de l’adénosine de type A1, est capable quant à lui d’induire une tolérance ischémique ou épileptique. Cela suggère que chacun des éléments intervenant dans l’établissement de la tolérance cérébrale représente une cible thérapeutique potentielle. crom : (-)cromakalim ; R-PIA : R(-)N6-(2-phénylisopropyl)-adénosine.
Les deux critères majeurs régissant cette tolérance sont l’intensité du premier stress ischémique et le délai séparant les deux agressions ischémiques. La protection est induite par une ischémie modérée, non létale pour les neurones, et apparaît uniquement si le délai entre les deux ischémies est de plusieurs dizaines heures [5]. La protection est transitoire et implique une fenêtre de protection de 24 à 96 heures après la première ischémie (Figure 1). La tolérance est établie par la quasi-totalité des agressions ischémiques, qu’elles soient focales (occlusion de l’artère cérébrale moyenne), globales (arrêt total du débit sanguin cérébral) ou croisées (pour revue, voir [6]). Des modèles de tolérance neuronale ischémique ont été développés in vitro [6]. Les conditions induisant l’ischémie sont reproduites en associant une hypoxie à un appauvrissement en glucose du milieu de culture. Ce modèle respecte le profil des dommages neuronaux observé in vivo et la tolérance ischémique observée semble régie par les mêmes critères.
La tolérance épileptique
Une des premières descriptions relatives à ce phénomène concerne deux modèles d’épilepsie chez l’animal : l’embrasement limbique par kindling dans le cortex pyriforme et l’injection d’un analogue du glutamate (le principal neuromédiateur excitateur), l’acide kaïnique. Son injection chez des rats, préalablement soumis à un embrasement limbique, n’induit pas les dommages neuronaux attendus avec ce type d’agression épileptique [7]. Depuis, la tolérance épileptique a été définie par analogie avec la tolérance ischémique [4] (Figures 1 et 2) et mise en évidence dans plusieurs modèles expérimentaux, avec différents types de crises épileptiques induites par différents neurotoxiques [4, 8]. Des études in vitro, notamment sur des cultures organotypiques d’hippocampe, ont confirmé que l’exposition préalable de neurones à une dose sublétale d’acide kaïnique les protège des dommages normalement induits par une dose toxique (voire létale) de cet agent [9].
La tolérance croisée et le préconditionnement pharmacologique
La définition des critères régissant la tolérance ischémique ou épileptique a ouvert de nouveaux champs d’investigation avec deux nouveaux aspects de la tolérance cérébrale : la tolérance croisée et le préconditionnement pharmacologique. La notion de tolérance croisée repose sur le fait que de nombreux stimulus supposés être nocifs, comme l’hyperthermie, la cortical spreading depression ou le stress oxydatif augmentent la résistance du cerveau à une agression cérébrale secondaire d’un type différent. Le premier stimulus, toujours sublétal, est perçu par les neurones comme un signal de stress.
Le préconditionnement pharmacologique implique que la molécule utilisée pour induire la résistance du cerveau stimule les mécanismes biologiques probablement mis en jeu lors de l’établissement de la tolérance cérébrale. Pour établir celle-ci, la réponse neuronale nécessite toujours un délai de mise en oeuvre associé à la synthèse de nouvelles protéines protectrices ou la répression de protéines de type pro-apoptotique [10].
La démonstration in vivo de l’existence de la tolérance croisée (ischémie/épilepsie, épilepsie/ischémie) [8, 11] et l’établissement de modèles de préconditionnement pharmacologique [10, 12, 13] offrent de nouvelles opportunités, tant dans le domaine de la prévention - au niveau clinique - qu’en recherche fondamentale, dans l’étude des mécanismes communs aux différents préconditionnements.
Les mécanismes classiques de la tolérance cérébrale
L’efficacité d’un préconditionnement est associée à la notion de seuil « à franchir pour protéger » et « à ne pas dépasser pour éviter la nocivité ». Elle repose sur l’activation de nombreux de facteurs, d’où sa complexité (Figure 3 et 4).
Figure 3
Où, quand et comment les différents composants cellulaires impliqués dans les fenêtres précoce et retardée de la tolérance cérébrale peuvent s’opposer à la cascade neurodestructrice induite par une agression cérébrale sévère.
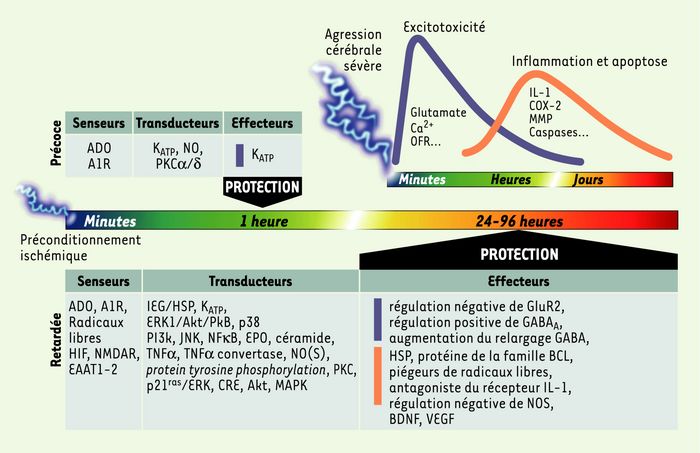
Une ischémie ou une crise épileptique sévère induisent, selon un profil temporel bien défini, une série complexe de mécanismes (excitotoxicité, apoptose, inflammation) qui aboutit au dommage cérébral. Le préconditionnement ischémique induit deux fenêtres de protection (précoce et retardée), dont les délais d’établissement sont différents et dont l’efficacité ne couvre pas la même échelle de temps. L’heure de protection offerte par la tolérance précoce s’oppose aux quelques jours (2 à 4) de la tolérance retardée. Cette composante temporelle explique pourquoi certains facteurs comme les senseurs (adénosine ou récepteur A1) interviennent avec le même rôle dans les deux types de protection, alors que d’autres ont un double rôle de transducteur ou d’effecteur (KATP). La résistance neuronale sera réelle si les effecteurs induits dans la fenêtre de protection s’opposent efficacement à la cascade de mécanismes délétères déclenchée par l’agression cérébrale sévère. Les multiples effecteurs de la protection retardée ont des actions anti-excitotoxique, anti-inflammatoire et anti-apoptotique, garantissant ainsi une protection maximale. A1R : adenosine receptor type1 ; ADO : adénosine ; Akt : a serine-threonine kinase ; BDNF : brain derived neutrophic factor ; COX-2 : cyclo-oxygénase ; CRE : cAMP-response element ; EAAT : excitatory amino acid transporter ; EPO : érythropoïétine ; ERK : extracellular signal-regulated kinase ; GluR2 : glutamate-receptor subunit 2 ; GABA : 4-aminobutyric acid ; GABAA : GABA-Areceptor ; HIF : hypoxia inducible factor ; HSP : heat shock protein ; IEG : immediate early gene ; IL-1 : interleukine-1 : JNK : c-jun N-terminal kinase ; KATP : ATP-sensitive potassium channel ; MAPK : mitogen-activated protein kinase ; MMP : matrix metalloproteases ; NF-κB : nuclear-factorκB ; NMDAR : NMDAreceptor ; NO : nitric oxide ; NOS : nitric oxide synthase ; OFR : oxygen free radical ; p21ras : p21 mitogen-activated protein kinase ; p38 : p38mitogen-activated protein kinase ; PI3K : phospho-inositide-3 kinase ; PKB : protéine kinase B ; PKC : protéine kinase C ; TNF : tumor necrosis factor ; VEGF : vascular-endothelial growth factor.
Figure 4
Mécanismes classiques de la tolérance cérébrale.

Les préconditionnements ischémique et épileptique activent les récepteurs NMDA (N-méthyl-D-aspartate) et les récepteurs de l’adénosine de type A1 qui - via une cascade d’activation impliquant des protéines G et la protéine kinase C (PKC) - ouvrent les canaux potassiques sensibles à l’ATP (KATP). Les voies de signalisation intracellulaire résultant de ces activations au niveau membranaire convergent vers l’activation de facteurs de transcription - comme le facteur nucléaire κB (NF-κB) - dont la translocation au niveau nucléaire induit la synthèse de protéines bénéfiques pour les neurones, par exemple la protéine de choc thermique HSP70. Cet ensemble de modifications biochimiques amène les neurones à un état de tolérance. Ce dernier définit la fenêtre de protection (retardée), pendant laquelle une agression cérébrale supposée délétère n’aurait en fait qu’une très faible incidence sur le cerveau. Les préconditionnements pharmacologiques par les activateurs des canaux KATP (récepteurs de type A1) et des récepteurs NMDA reproduisent certaines des voies de signalisation des préconditionnements dits « classiques », comme le montrent l’activation du facteur NF-κB et la synthèse de la protéine HSP70. Leur fenêtre de protection est identique à celle induite par les préconditionnements ischémique et épileptique. MnSOD : manganese superoxide dismutase ; iNOS : inducible nitric oxide synthase.
Parmi les différents composants cellulaires, trois catégories très liées entre elles peuvent être distinguées : les « senseurs, ou initiateurs », qui capturent le signal de stress et activent une multitude de « voies de signalisation intracellulaire, transducteurs membranaires et intracellulaires » qui eux-mêmes convergent vers des « effecteurs » de la protection (pour revue, voir [14]). La plupart de ces facteurs sont communs à tous les modèles de préconditionnement in vivo et in vitro, et leur polyvalence est à l’origine de la protection croisée. Chacun d’entre eux représente une cible thérapeutique potentielle dont l’activation pourrait améliorer la prise en charge des patients victimes d’accidents ischémiques ou épileptiques.
La réponse au stress est dans une large mesure identique pour tous les types cellulaires. Les neurones possèdent cependant des voies de signalisation intracellulaire spécifiques. Une des distinctions les plus marquantes entre les neurones et les autres cellules existe au niveau des senseurs, ou initiateurs, et implique le glutamate et ses récepteurs.
Le glutamate et ses récepteurs
Pour l’ischémie et l’épilepsie, l’hyperexcitabilité et la mort neuronales sont associées à la libération excessive du neurotransmetteur excitateur, le glutamate. Le préconditionnement n’affecte pas la libération du glutamate au cours d’une ischémie sévère [15], mais prépare les neurones à résister à son action toxique (excitotoxicité) : il agit sur les récepteurs du glutamate. In vitro, un préconditionnement pharmacologique par l’activation des récepteurs NMDA (N-méthyl-D-aspartate) établit une tolérance neuronale identique à celle obtenue par le préconditionnement ischémique ou épileptique [16]. Le préconditionnement prévient l’altération induite par l’ischémie sévère de la sous-unité GluR2 des récepteurs AMPA (α-amino-3hydroxy-5-méthyl-4-isoxazolepropionate), partenaire de la résistance à l’excitotoxicité [17].
Les récepteurs de l’adénosine de type A1 et les canaux potassiques sensibles à l’ATP (KATP)
Ces protéines membranaires sont un des sujets les plus documentés de la tolérance cérébrale. Un préconditionnement pharmacologique par les activateurs des canaux KATP ou des récepteurs de type A1 reproduit une fenêtre de protection identique à celle observée dans la tolérance ischémique ou épileptique, et déclenche un signal de stress conduisant à la synthèse de nombreuses protéines, dont HSP70 (heat shock protein), décrite pour ses effets bénéfiques dans la majorité des modèles de préconditionnement [10]. L’inhibition des canaux KATP ou des récepteurs de type A1 abolit les effets bénéfiques induits par un préconditionnement ischémique ou épileptique [17], tandis que leur activation en phase aiguë protège les neurones d’une ischémie ou d’une crise épileptique [18]. Les récepteurs A1 de l’adénosine et les canaux KATP pourraient donc avoir à la fois un rôle de senseur et d’effecteur dans la tolérance cérébrale, et l’analyse de leur rôle dans la phase tardive du préconditionnement cérébral est aussi compliquée par l’existence potentielle de canaux KATP mitochondriaux [19].
Le facteur nucléaire κB (NF-κB)
Le rôle des facteurs de transcription comme NF-κB n’a été étudié que très récemment, lorsque le délai nécessaire à l’établissement de la tolérance cérébrale a clairement été associé à la synthèse de nouvelles protéines. Ils représentent un intérêt particulier puisqu’ils pourraient être un des points de convergence obligatoire à tous les préconditionnements. Le facteur NF-κB a retenu l’attention, car il est activé par de nombreux stimulus (anoxie, radicaux libres, ischémie…). On lui attribue de plus un double rôle : pro- et anti-apoptotique [20]. Cette dualité en accord avec le concept du préconditionnement a motivé son étude dans la tolérance cérébrale. Il apparaît aujourd’hui comme un des points de convergence clé des nombreuses voies de signalisation intracellulaire déclenchées aussi bien par les préconditionnements classiques (ischémique et épileptique) que par les préconditionnements pharmacologiques (activateurs de canaux KATP, récepteurs A1 ou prétraitement par les acides gras poly-insaturés). Dans tous ces modèles, l’établissement de la tolérance est aboli si l’activation du facteur NF-κB est inhibée [21].
Conclusions
Les quelques exemples évoqués montrent que de nombreux modèles de tolérance cérébrale ont été caractérisés, y compris chez l’homme [22, 23], suscitant un intérêt clinique grandissant. Les cinétiques d’action sont connues. Les études ont permis d’élargir la notion de tolérance à la capacité de résistance intrinsèque des neurones face à des agressions délétères et la notion de préconditionnement à une multitude de stress cellulaires. L’existence de mécanismes endogènes communs aux différents préconditionnements ne fait maintenant plus aucun doute. L’approche génomique, grâce aux nouvelles techniques d’analyse génomique et protéomique, apporte un outil supplémentaire à la caractérisation de la réponse aux différents préconditionnements. Les différentes approches devraient pouvoir aboutir à une nouvelle stratégie thérapeutique fondée sur le préconditionnement pharmacologique, par exemple dans le cas de la chirurgie cérébrale à haut risque de complications vasculaires ou épileptiques.
Parties annexes
Remerciements
Nous tenons vivement à remercier les professeurs M. Lazdunski et M. Moskovitz de leur confiance et leurs encouragements, ainsi que le docteur C. Waeber pour sa lecture attentive du manuscrit, sans oublier le CNRS et la Fondation de la Recherche Médicale pour leur soutien financier.
Références
- 1. Murry CE, Jennings R, Reimer K. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 1986 ; 74 : 1124-36.
- 2. Schurr A, Reid KH, Tseng MT, et al. Adaptation of adult brain tissue to anoxia and hypoxia in vitro. Brain Res 1986 ; 374 : 244-8.
- 3. Kitagawa K, Matsumoto M, Tagaya M, et al. « Ischemic tolerance » phenomenon found in the brain. Brain Res 1990 ; 528 : 21-4.
- 4. Sasahira M, Lowry T, Simon RP, Greenberg DA. Epileptic tolerance: Prior seizures protect against seizure-induced neuronal injury. Neurosci Lett 1995 ; 185 : 95-8.
- 5. Kato H, Liu Y, Araki T, Kogure K. Temporal profile of the effects of pretreatment with brief cerebral ischemia on the neuronal damage following secondary ischemic insult in the gerbil: cumulative damage and protective effects. Brain Res 1991 ; 553 : 238-42.
- 6. Schaller B, Graf R. Cerebral ischemic preconditioning. An experimental phenomenon or a clinical important entity of stroke prevention? J Neurol 2002 ; 249 : 1503-11.
- 7. Kelly ME, McIntyre DC. Hippocampal kindling protects several structures from the neuronal damage from kainic acid-induced status epilepticus. Brain Res 1994 ; 634 : 245-56.
- 8. Plamondon H, Blondeau N, Heurteaux C, Lazdunski M. Mutually protective actions of kainic acid epileptic preconditioning and sublethal global ischemia on hippocampal neuronal death: Involvement of adenosine A1 receptors and KATP channels. J Cereb Blood Flow Metab 1999 ; 19 : 1296-308.
- 9. Best N, Sundstrom LE, Mitchell J, Wheal HV. Pre-exposure to subtoxic levels prevents kainic acid lesions in organotypic hippocampal slice cultures: effects of kainic acid on parvalbumin-immunoreactive neurons and expression of heat shock protein 72 following the induction of tolerance. Eur J Neurosci 1996 ; 8 : 1209-19.
- 10. Blondeau N, Plamondon H, Richelme C, et al. KATP channel openers, adenosine agonists and epileptic preconditioning are stress signals inducing hippocampal neuroprotection. Neuroscience 2000 ; 100 : 465-74.
- 11. Pohle W, Rauca C. Hypoxia protects against the neurotoxicity of kainic acid. Brain Res 1994 ; 644 : 297-304.
- 12. Blondeau N, Widmann C, Lazdunski M, Heurteaux C. Polyunsaturated fatty acids induce ischemic and epileptic tolerance. Neuroscience 2001 ; 109 : 231-41.
- 13. Blondeau N, Lauritzen I, Widmann C, et al. A potent protective role of lysophospholipids against global cerebral ischemia and glutamate excitotoxicity in neuronal cultures. J Cereb Blood Flow Metab 2002 ; 22 : 821-34.
- 14. Dirnagl U, Simon RP, Hallenbeck JM. Ischemic tolerance and endogenous neuroprotection. Trends Neurosci 2003 ; 26 : 248-54.
- 15. Nakata N, Kato H, Liu Y, Kogure K. Effects of pretreatment with sublethal ischemia on the extracellular glutamate concentrations during secondary ischemia in the gerbil hippocampus evaluated with intracerebral microdialysis. Neurosci Lett 1992 ; 138 : 86-8.
- 16. Grabb MC, Choi DW. Ischemic tolerance in murine cortical cell culture: critical role for NMDA receptors. J Neurosci 1999 ; 19 : 1657-62.
- 17. Heurteaux C, Lauritzen I, Widmann C, Lazdunski M. Essential role of adenosine, adenosine A1 receptors and KATP channels in cerebral ischemic preconditioning. Proc Natl Acad Sci USA 1995 ; 92 : 4666-70.
- 18. Heurteaux C, Bertaina V, Widmann C, Lazdunski M. K+ channel openers prevent global ischemia-induced expression of c-fos, c-jun, heat shock protein, and amyloid β-protein precursor genes and neuronal death in rat hippocampus. Proc Natl Acad Sci USA 1993 ; 90 : 9431-5.
- 19. Liu Y, Sato T, Seharaseyon J, et al. Mitochondrial ATP-dependent potassium channels. Viable candidate effectors of ischemic preconditioning. Ann NY Acad Sci 1999 ; 874 : 27-37.
- 20. Mattson MP, Culmsee C, Yu Z, Camandola S. Roles of nuclear factor kappaB in neuronal survival and plasticity. J Neurochem 2000 ; 74 : 443-56.
- 21. Blondeau N, Widmann C, Lazdunski M, Heurteaux C. Activation of the nuclear factor-kappaB is a key event in brain tolerance. J Neurosci 2001 ; 21 : 4668-77.
- 22. Moncayo J, de Freitas GR, Bogousslavsky J, et al. Do transient ischemic attacks have a neuroprotective effect? Neurology 2000 ; 54 : 2089-94.
- 23. Weih M, Kallenberg K, Bergk A, et al. Attenuated stroke severity after prodromal TIA: A role for ischemic tolerance in the brain? Stroke 1999 ; 30 : 1851-4.
Liste des figures
Figure 1
Critères définissant les concepts du préconditionnement et de la tolérance cérébrale.
Il existe plusieurs types de préconditionnement cérébral, définis en fonction de leur inducteur (la première agression cérébrale, qui doit dans tous les cas être modérée). Il existe toujours un délai, impliquant des changements biochimiques, avant que la fenêtre de protection ne s’établisse. Les deux fenêtres de protection généralement décrites sont transitoires et ont des profils temporels distincts : la fenêtre précoce est de courte durée et apparaît rapidement après le préconditionnement ; la fenêtre retardée est quant à elle effective entre 24 et 96 heures après le préconditionnement.
Figure 2
Protection des neurones de l’hippocampe par différents types de préconditionnement et analogie entre les tolérances ischémique et épileptique.
La tolérance cérébrale ischémique décrite dans cette figure est induite par un préconditionnement ischémique (ischémie cérébrale modérée). Ce dernier réduit la neurodégénérescence au niveau des couches du CA1 de l’hippocampe induite par une ischémie cérébrale sévère lorsque le cerveau n’est pas préconditionné. De même, la tolérance épileptique est induite par un préconditionnement épileptique (crise épileptique modérée) qui préserve les neurones des couches du CA3 de l’hippocampe détruits lors d’une crise épileptique sévère. Le préconditionnement pharmacologique, par les activateurs des canaux KATP ou des récepteurs de l’adénosine de type A1, est capable quant à lui d’induire une tolérance ischémique ou épileptique. Cela suggère que chacun des éléments intervenant dans l’établissement de la tolérance cérébrale représente une cible thérapeutique potentielle. crom : (-)cromakalim ; R-PIA : R(-)N6-(2-phénylisopropyl)-adénosine.
Figure 3
Où, quand et comment les différents composants cellulaires impliqués dans les fenêtres précoce et retardée de la tolérance cérébrale peuvent s’opposer à la cascade neurodestructrice induite par une agression cérébrale sévère.
Une ischémie ou une crise épileptique sévère induisent, selon un profil temporel bien défini, une série complexe de mécanismes (excitotoxicité, apoptose, inflammation) qui aboutit au dommage cérébral. Le préconditionnement ischémique induit deux fenêtres de protection (précoce et retardée), dont les délais d’établissement sont différents et dont l’efficacité ne couvre pas la même échelle de temps. L’heure de protection offerte par la tolérance précoce s’oppose aux quelques jours (2 à 4) de la tolérance retardée. Cette composante temporelle explique pourquoi certains facteurs comme les senseurs (adénosine ou récepteur A1) interviennent avec le même rôle dans les deux types de protection, alors que d’autres ont un double rôle de transducteur ou d’effecteur (KATP). La résistance neuronale sera réelle si les effecteurs induits dans la fenêtre de protection s’opposent efficacement à la cascade de mécanismes délétères déclenchée par l’agression cérébrale sévère. Les multiples effecteurs de la protection retardée ont des actions anti-excitotoxique, anti-inflammatoire et anti-apoptotique, garantissant ainsi une protection maximale. A1R : adenosine receptor type1 ; ADO : adénosine ; Akt : a serine-threonine kinase ; BDNF : brain derived neutrophic factor ; COX-2 : cyclo-oxygénase ; CRE : cAMP-response element ; EAAT : excitatory amino acid transporter ; EPO : érythropoïétine ; ERK : extracellular signal-regulated kinase ; GluR2 : glutamate-receptor subunit 2 ; GABA : 4-aminobutyric acid ; GABAA : GABA-Areceptor ; HIF : hypoxia inducible factor ; HSP : heat shock protein ; IEG : immediate early gene ; IL-1 : interleukine-1 : JNK : c-jun N-terminal kinase ; KATP : ATP-sensitive potassium channel ; MAPK : mitogen-activated protein kinase ; MMP : matrix metalloproteases ; NF-κB : nuclear-factorκB ; NMDAR : NMDAreceptor ; NO : nitric oxide ; NOS : nitric oxide synthase ; OFR : oxygen free radical ; p21ras : p21 mitogen-activated protein kinase ; p38 : p38mitogen-activated protein kinase ; PI3K : phospho-inositide-3 kinase ; PKB : protéine kinase B ; PKC : protéine kinase C ; TNF : tumor necrosis factor ; VEGF : vascular-endothelial growth factor.
Figure 4
Mécanismes classiques de la tolérance cérébrale.
Les préconditionnements ischémique et épileptique activent les récepteurs NMDA (N-méthyl-D-aspartate) et les récepteurs de l’adénosine de type A1 qui - via une cascade d’activation impliquant des protéines G et la protéine kinase C (PKC) - ouvrent les canaux potassiques sensibles à l’ATP (KATP). Les voies de signalisation intracellulaire résultant de ces activations au niveau membranaire convergent vers l’activation de facteurs de transcription - comme le facteur nucléaire κB (NF-κB) - dont la translocation au niveau nucléaire induit la synthèse de protéines bénéfiques pour les neurones, par exemple la protéine de choc thermique HSP70. Cet ensemble de modifications biochimiques amène les neurones à un état de tolérance. Ce dernier définit la fenêtre de protection (retardée), pendant laquelle une agression cérébrale supposée délétère n’aurait en fait qu’une très faible incidence sur le cerveau. Les préconditionnements pharmacologiques par les activateurs des canaux KATP (récepteurs de type A1) et des récepteurs NMDA reproduisent certaines des voies de signalisation des préconditionnements dits « classiques », comme le montrent l’activation du facteur NF-κB et la synthèse de la protéine HSP70. Leur fenêtre de protection est identique à celle induite par les préconditionnements ischémique et épileptique. MnSOD : manganese superoxide dismutase ; iNOS : inducible nitric oxide synthase.