Corps de l’article
L’AMP-activated protein kinase (AMPK), enzyme ubiquitaire, est proposée comme le détecteur métabolique de la cellule lui permettant de s’adapter aux modifications de son environnement [1]. L’AMPK est un complexe trimérique constitué d’une sous-unité catalytique α et de deux sous-unités régulatrices β et γ. Il existe deux isoformes α (α1 et α2), deux isoformes β (β1 et β2) et trois isoformes γ (γ1, γ2, γ3) [1]. Chacune de ces isoformes est codée par un gène différent. L’AMPK est activée physiologiquement par l’augmentation de la concentration intracellulaire en AMP lors des situations de carence énergétique cellulaire (hypoxie, hypoglycémie et exercice physique) [2]. L’activation de l’AMPK réduit les voies métaboliques consommatrices d’énergie (comme la lipogenèse ou la synthèse stéroïdienne) et augmente les voies productrices d’ATP (comme l’oxydation des acides gras). Grâce au 5-amino-imidazolecarboxamide riboside (AICAR), analogue non métabolisable de l’AMP capable d’activer l’AMPK, les rôles de l’AMPK ont pu être peu à peu précisés in vitro [2] et in vivo [3, 4]. Dans le muscle, l’activation de l’AMPK stimule l’augmentation du captage insulino-indépendant du glucose [2] et l’oxydation des acides gras [2]. On connaît le rôle de la leptine dans l’augmentation de l’oxydation musculaire des acides gras, mais cet effet nécessite l’activation de l’AMPK ((→) m/s 1999, n° 11, p. 1276) [5]. Dans le foie, l’activation de l’AMPK par l’AICAR diminue la néoglucogenèse in vitro [6] et la production hépatique de glucose in vivo [3, 6]. Les effets métaboliques de l’AMPK que nous venons de décrire dépendent surtout de l’isoforme catalytique α2 de l’AMPK [2]. Ainsi, en stimulant le captage de glucose insulino-indépendant et l’oxydation lipidique des acides gras dans le muscle, et en diminuant la production hépatique de glucose, l’activation de l’AMPK pourrait avoir des effets bénéfiques sur les anomalies métaboliques observées au cours du diabète de type 2 et de l’obésité [2].
Pour mieux connaître les rôles physiologiques de l’isoforme catalytique α2 de l’AMPK sans avoir à utiliser de moyens pharmacologiques comme l’AICAR, nous avons créé au laboratoire un modèle de rongeurs dont le gène codant pour cette sous-unité a été invalidé [7].
Les souris AMPKα2-/- n’ont pas d’anomalies apparentes. Ces souris ont, après un jeûne court, une glycémie et une insulinémie similaires à celles des souris témoins. Lorsque l’apport nutritif n’est pas limité, et lors d’une hyperglycémie provoquée par voie intrapéritonéale, on observe chez les souris mutantes une hyperglycémie associée à une sécrétion réduite d’insuline. De manière surprenante, la sécrétion d’insuline chez ces animaux, mesurée après exposition in vitro d’îlots pancréatiques isolés à une large gamme de concentrations de glucose (de 3 à 25 mM), est normale. Par ailleurs, le contenu en insuline et l’expression du gène de l’insuline dans les cellules β pancréatiques sont similaires chez les souris AMPKα2-/- et les souris témoins. Ces observations suggèrent fortement que le défaut de sécrétion d’insuline observé in vivo chez les souris AMPKα2-/- est lié à un défaut de régulation de la sécrétion d’insuline en situation de charge en glucose. Nous avons pu exclure des facteurs dont on sait qu’ils altèrent le contrôle de la sécrétion d’insuline chez le rongeur comme l’hyperleptinémie, l’accumulation d’acides gras dans la cellule β pancréatique ou l’hypokaliémie. L’équilibre entre les tonus sympathique et parasympathique est connu pour être un des facteurs clés de la régulation de la sécrétion d’insuline in vivo. Pour déterminer si une altération du système nerveux autonome explique les anomalies des souris AMPKα2-/-, nous avons traité ces souris par un antagoniste des récepteurs α-adrénergiques. Ce traitement a normalisé la cinétique de l’hyperglycémie des souris AMPKα2-/- provoquée par voie intrapéritonéale. Ce résultat, associé à l’existence d’une excrétion urinaire accrue en catécholamines, démontre que l’exagération du tonus sympathique est bien responsable du défaut de sécrétion d’insuline observé chez les souris AMPKα2-/-.
La technique du clamp euglycémique hyper-insulinémique[*] révèle par ailleurs que les souris AMPKα2-/- présentent une résistance à l’insuline au niveau musculaire. Cette résistance est secondaire à un défaut de synthèse de glycogène dans le muscle squelettique au cours du clamp. Le tonus sympathique exagéré est-il suffisant pour expliquer la résistance à l’insuline des souris AMPKα2-/-? Puisque le clamp étudie la sensibilité à l’insuline du muscle squelettique, ne pouvait-on pas plutôt expliquer cette résistance par l’absence de la sous-unité α2 dans le muscle? Pour le savoir, nous avons étudié la sensibilité à l’insuline d’un modèle de dominant négatif musculaire de l’AMPK. La sensibilité à l’insuline normale dans ce modèle suggère que l’absence d’activité AMPK dans le muscle ne peut pas à elle seule expliquer le phénotype observé [7].
Les souris AMPKα2-/- présentent donc des anomalies métaboliques in vivo (réduction de la sécrétion de l’insuline et résistance à l’hormone, défaut de synthèse de glycogène musculaire) qui peuvent être expliquées par un tonus sympathique exagéré. Ces anomalies métaboliques n’existent qu’en période de charge en glucose (hyperglycémie provoquée, apport de nourriture, clamp euglycémique) et sont indétectables à jeun. On sait que le cerveau (en particulier l’hypothalamus) intègre les signaux métaboliques périphériques pour modifier en retour, grâce au système nerveux autonome, la sécrétion d’insuline et la sensibilité du muscle à cette hormone. Ainsi, nos travaux montrent que l’absence de la sous-unité α2 de l’AMPK dans le cerveau provoque une exagération du tonus sympathique qui altère le métabolisme des organes périphériques (Figure 1). Il faut donc inclure la sous-unité α2 de l’AMPK à la liste des effecteurs contrôlant le métabolisme via le système nerveux autonome.
Figure 1
Impact sur le métabolisme périphérique de l’absence de la sous-unité α2 de l’AMPK.
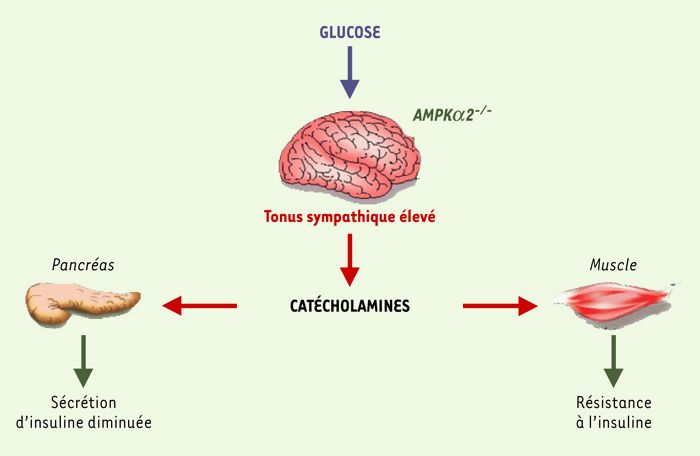
L’absence de la sous-unité α2 de l’AMPK dans le cerveau altère le tonus sympathique, ce qui provoque la réduction de la sécrétion d’insuline et de la sensibilité à l’insuline du muscle. La sous-unité α2 de l’AMPK cérébrale participe au contrôle du métabolisme périphérique en modulant principalement l’activité du système nerveux autonome.
Parties annexes
Notes
-
[*]
Il s’agit d’une technique classique de mesure des effets métaboliques de l’insuline: pratiquement, l’insuline est administrée in vivo à un taux constant pendant 3-4 h, et le taux de glucose est maintenu stable par l’injection concomitante de glucose. Cela permet l’évaluation des effets de l’insuline indépendamment de son effet sur la glycémie.
Références
- 1. Hardie DG, Carling D, Carlson M. The AMP-activated/SNF1protein kinase subfamily: metabolic sensors of the eukaryotic cell? Annu Rev Biochem 1998; 67: 821-55.
- 2. Winder WW, Hardie DG. AMP-activated protein kinase, a metabolic master switch: possible roles in type 2 diabetes. Am J Physiol 1999; 277: E1-10.
- 3. Bergeron R, Previs SF, Cline GW, et al. Effect of 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside infusion on in vivo glucose and lipid metabolism in lean and obese Zucker rats. Diabetes 2001; 50: 1076-82
- 4. Bergeron R, Russell RR, Young LH, et al. Effect of AMPK activation on muscle glucose metabolism in conscious rats. Am J Physiol 1999; 276: E938-44.
- 5. Minokoshi Y, Kim YB, Peroni OD, et al. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 2002; 415: 339-43.
- 6. Zhou G, Myers R, Li Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 2001; 108: 1167-74.
- 7. Viollet B, Andreelli F, Jorgensen SB, et al. The AMP-activated protein kinase α2 catalytic subunit controls whole body insulin sensitivity. J Clin Invest 2003; 111: 91-8.
Liste des figures
Figure 1
Impact sur le métabolisme périphérique de l’absence de la sous-unité α2 de l’AMPK.
L’absence de la sous-unité α2 de l’AMPK dans le cerveau altère le tonus sympathique, ce qui provoque la réduction de la sécrétion d’insuline et de la sensibilité à l’insuline du muscle. La sous-unité α2 de l’AMPK cérébrale participe au contrôle du métabolisme périphérique en modulant principalement l’activité du système nerveux autonome.