Corps de l’article
Le prion est l’agent transmissible responsable d’encéphalopathies spongiformes chez plusieurs espèces de mammifères: l’ESB chez les bovins, la maladie de Creutzfeldt-Jakob chez les humains, la tremblante (scrapie) chez le mouton, ou encore la cachexie chronique (ou syndrome de dégénérescence chronique, chronic wasting disease, CWD) du cerf et de l’élan. La maladie est également observée chez des espèces moins communes, telles que certains ongulés exotiques et des félins en captivité [1].
Les prions sont vraisemblablement des protéines, légèrement différentes d’une espèce à une autre, qui peuvent adopter au moins deux conformations spatiales, une conformation cellulaire «saine» (PrPC), présente de façon ubiquitaire dans l’organisme, et une conforma-tion pathogène (PrPSc). Contrairement à un virus, qui se multiplie dans les cellules de son hôte grâce à son patrimoine génétique (constitué d’acides nucléiques, ADN ou ARN), la protéine PrPSc anormale semble se propager au sein de l’organisme, et d’un animal à un autre, en convertissant en molécules pathogènes les protéines du prion saines avec lesquelles elle entre en contact [2]. Toutefois, plus de vingt ans après sa formulation initiale par Stanley Prusiner, cette hypothèse de la protéine seule (protein-only hypothesis) suscite encore une foule d’interrogations malgré l’apport de plusieurs éléments de réponse ces dernières années. En particulier, la protéine prion de mammifère, PrPC a été convertie in vitro en une entité dotée de propriétés physico-chimiques caractéristiques de la protéine prion pathogène, PrPSc [3]. Malgré tout, une des étapes cruciales concernant l’interaction entre les deux isoformes n’avait toujours pas été formellement démontrée in vivo.
Nous avons tenté d’apporter de nouveaux éléments en faveur de cette hypothèse [4]. La protéine prion de souris a été fusionnée avec le fragment Fc des immunoglobulines G humaines pour créer une molécule dimérique soluble et sécrétée (PrP-Fc2), contrairement à PrPC qui est normalement ancrée à la partie externe de la membrane cellulaire. Nous avons ensuite créé des souris transgéniques qui expriment cette molécule PrP-Fc2.
Les souris qui n’expriment pas PrPC ne sont pas susceptibles aux maladies du prion (scrapie) [5]. Dans un premier temps, nous avons croisé nos souris transgéniques PrP-Fc2 avec des souris qui n’ont pas le gène codant pour PrP, de manière à n’exprimer que la forme dimérique soluble (PrP-Fc2) sans la forme endogène PrPC. Les souris transgéniques exprimant le dimère PrP soluble et inoculées avec des prions ne développèrent pas la scrapie et n’accumulèrent pas PrPSc, suggérant que PrP-Fc2 ne pouvait pas être convertie en une forme anormale PrP-FcSc(Figure 1B).
Figure 1
Un modèle pour l’action anti-prion de PrP-Fc2.
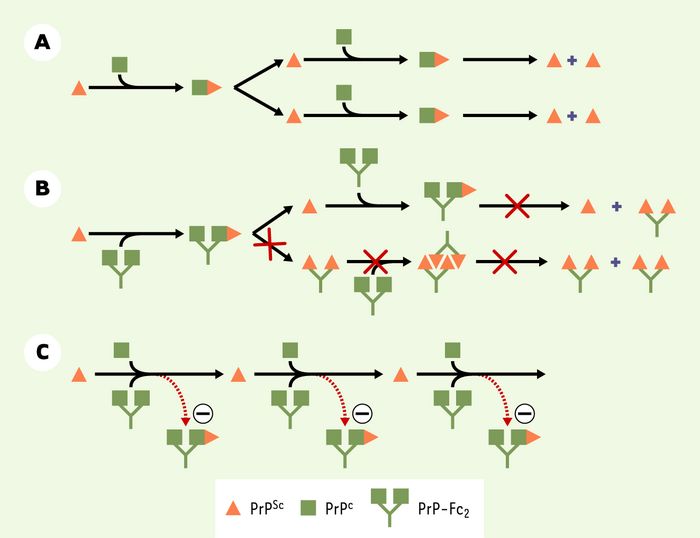
A. Selon le modèle dit du «redéploiement» (nb) PrPSc interagit avec PrPC de manière à former un dimère transitoire. PrPC est transformé en une nouvelle molécule PrPSc. La conversion spontanée dans le sens inverse (PrPSc en PrPc) serait rendue impossible en raison d’une barrière énergétique très élevée. B. En l’absence de PrPC, PrP-Fc2 ne permet ni la réplication de l’agent infectieux, ni la formation de PrPSc. Les souris ne développent ainsi aucun symptôme. C. Les souris co-exprimant PrPC endogène et PrP-Fc2 répliquent des prions et développent la scrapie. Plusieurs expériences suggèrent que PrP-Fc2 séquestre PrPSc et la rend incapable de convertir de nouvelles molécules PrPC en PrPSc, ce qui expliquerait pourquoi les souris transgéniques développent la scrapie, mais avec une cinétique ralentie.
Ces souris ont ensuite été croisées avec des souris sauvages de manière à exprimer simultanément PrP soluble et PrPC endogène. L’inoculation intracérébrale de prions a entraîné le développement de la maladie mais avec un retard significatif: 170 jours pour les souris témoins contre 280 jours pour les souris mutantes. Plusieurs animaux sacrifiés pendant la progression de la maladie révélèrent une diminution de l’accumulation de la forme anormale PrPSc, ainsi qu’une réplication ralentie de l’agent infectieux.
Un des éléments centraux de l’hypothèse de la protéine seule repose sur une interaction entre la forme normale PrPC et la forme anormale PrPSc(Figure 1A). La forme soluble PrP-Fc2 (qui n’est pas convertible en une forme pathogène) interagirait-elle avec la forme anormale, bloquant ainsi la propagation de PrPSc ?
C’est ce que nous avons ensuite essayé de tester en utilisant diverses méthodes biochimiques. Pour cela, nous avons couplé des billes magnétiques avec des anticorps anti-Fc humains ou avec la protéine A. Ces deux molécules capturent le fragment Fc, et donc la protéine dimérique PrP-Fc2, mais pas PrP seule. Les billes couplées ont ensuite été incubées avec des homogénats de cerveaux de souris PrP-Fc2 ou sauvages, toutes deux infectées avec des prions. L’analyse par Western blot des immunoprécipitations a révélé la présence de PrPSc (résistant à la digestion de la protéinase K) seulement dans le cas où les cerveaux des souris PrP-Fc2 ont été utilisés, suggérant que PrPSc et PrP-Fc2 s’associent in vivo.
Ces résultats ont été confirmés en utilisant une approche expérimentale différente. PrPSc, de même que PrPC, sont attachées aux radeaux lipidiques (rafts), régions membranaires riches en lipides. Après une ultracentrifugation dans un gradient Nycodenz, PrPSc est détectée dans les fractions légères. Lorsque le même gradient a été appliqué à PrP-Fc2, celui-ci était présent dans les fractions lourdes du gradient, ce qui est le comportement attendu d’une protéine soluble. En revanche, lorsque nous avons appliqué la même approche aux souris PrP-Fc2 infectées avec des prions, les molécules PrP-Fc2 étaient détectées en haut du gradient, dans les mêmes fractions que PrPSc ! Nous avons donc conclu que la forme soluble PrP-Fc2 interagissait avec la forme anormale PrPSc.
Que se passe-t-il chez les souris PrP-Fc2 qui développent la scrapie plus tardivement que les témoins ? PrP-Fc2 se lie probablement avec PrPSc et empêche cette dernière de convertir PrPC en une nouvelle molécule anormale. La cinétique de conversion est ainsi ralentie et les souris succombent à la maladie plus tardivement (Figure 1C). Cela suggère que l’interaction entre les deux isoformes est requise pour la réplication de PrPScin vivo et confirme donc un des postulats centraux de l’hypothèse de la protéine seule.
Parties annexes
Références
- 1. Aguzzi A, Montrasio F, Kaeser PS. Prions: health scare and biological challenge. Nat Rev Mol Cell Biol 2001; 2:118-26.
- 2. Aguzzi A, Weissmann C. Prion research: the next frontiers. Nature 1997; 389: 795-8.
- 3. Kocisko DA, Come JH, Priola SA, et al. Cell-free formation of protease-resistant prion protein. Nature 1994; 370: 471-4.
- 4. Meier P, Genoud N, Prinz M, et al. Soluble dimeric prion protein binds PrPScin vivo and antagonizes prion disease. Cell 2003; 113: 49-60.
- 5. Büeler HR, Aguzzi A, Sailer A, et al. Mice devoid of PrP are resistant to scrapie. Cell 1993; 73:1339-47.
- 6. Fischer MB, Roeckl C, Parizek P, Schwarz HP, Aguzzi A. Binding of disease-associated prion protein to plasminogen. Nature 2000; 408: 479-83.
Liste des figures
Figure 1
Un modèle pour l’action anti-prion de PrP-Fc2.
A. Selon le modèle dit du «redéploiement» (nb) PrPSc interagit avec PrPC de manière à former un dimère transitoire. PrPC est transformé en une nouvelle molécule PrPSc. La conversion spontanée dans le sens inverse (PrPSc en PrPc) serait rendue impossible en raison d’une barrière énergétique très élevée. B. En l’absence de PrPC, PrP-Fc2 ne permet ni la réplication de l’agent infectieux, ni la formation de PrPSc. Les souris ne développent ainsi aucun symptôme. C. Les souris co-exprimant PrPC endogène et PrP-Fc2 répliquent des prions et développent la scrapie. Plusieurs expériences suggèrent que PrP-Fc2 séquestre PrPSc et la rend incapable de convertir de nouvelles molécules PrPC en PrPSc, ce qui expliquerait pourquoi les souris transgéniques développent la scrapie, mais avec une cinétique ralentie.