Résumés
Résumé
Les histones conventionnelles H2A, H2B, H3 et H4 sont des protéines basiques très conservées autour desquelles s’enroule l’ADN pour former l’unité de base de la chromatine: le nucléosome. Ces histones du coeur du nucléosome peuvent être remplacées par des variants qui sont présents chez tous les organismes eucaryotes. Les variants d’histones participent, avec les autres voies de modification du nucléosome, à la spécialisation fonctionnelle de la chromatine. Dans cette synthèse, nous nous focalisons sur trois variants majeurs de l’histone H2A (H2A.X, H2A.Z et macroH2A). Les recherches récentes mettent en lumière leur implication dans des évènements cruciaux comme la réparation de l’ADN ou la régulation de la transcription.
Summary
The histones H2A, H2B, H3 and H4 are very conserved basic proteins that wrap almost two turns of DNA to form the nucleosome core. Conventional histones can be replaced with histone variants that are found in all eukaryotic organisms. Together with other nucleosome modification pathways, histone variants participate in the functional specialization of chromatin. In this review, we focus on three major H2A histone variants: H2A.X, H2A.Z and macroH2A. Recent discoveries highlight their involvement in crucial events such as DNA repair and transcriptional regulation.
Corps de l’article
Dans le noyau des cellules eucaryotes, la compaction physique de l’ADN et la régulation de ses différentes fonctions sont assurées par des protéines spécifiques. Une catégorie de ces protéines, les histones H2A, H2B, H3 et H4, s’associent pour former un octamère autour duquel s’enroule l’ADN. La particule nucléo-protéique constituée est le nucléosome (Figure 1A). Le nucléosome est le constituant de base de la chromatine et joue un rôle essentiel dans les grandes fonctions nucléaires (transcription, réplication, réparation de l’ADN). Pour moduler son activité, la cellule met en oeuvre trois voies principales: le recrutement de complexes de remodelage du nucléosome, les modifications post-traductionnelles des histones et l’incorporation des variants d’histones (Figure 2).
Figure 1
Structure schématique de la particule coeur du nucléosome.
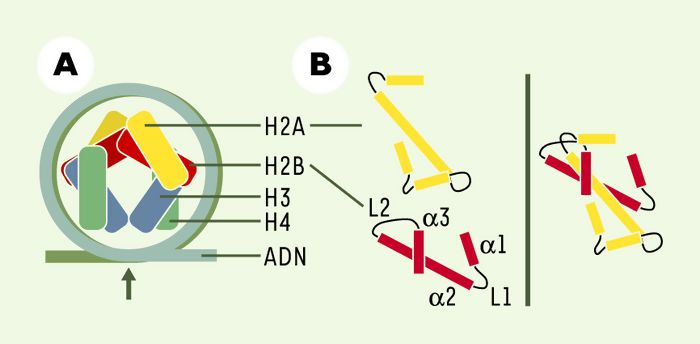
A. La double hélice d’ADN s’enroule autour de l’octamère d’histones. Les extrémités amino-terminales des histones ne sont pas représentées. B. Le repliement (histone fold) des hélices α et des boucles de connexion (L) des histones H2A et H2B permet leur dimérisation.
Figure 2
Les trois voies de modification du nucléosome.
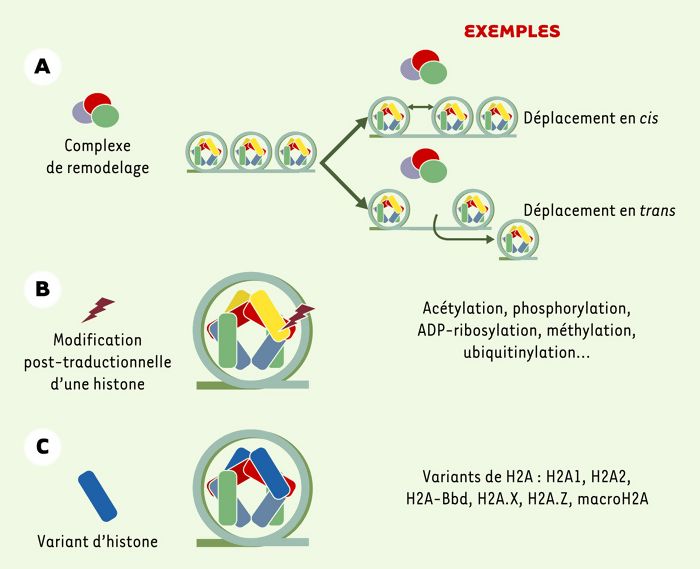
A. Déplacement en cis ou en trans des nucléosomes par un complexe de remodelage. B. Modification post-traductionnelle des extrémités des histones. C. Substitution d’une histone conventionnelle par un variant.
La première voie implique trois grandes familles de complexes de remodelage dépendants de l’ATP: SWI/SNF, ISW et Mi-2/NuRD. Toutes ont en commun la présence d’une sous-unité ATPase, et requièrent l’hydrolyse de l’ATP pour modifier la structure chromatinienne, par déplacement de l’octamère d’histones en cis (glissement) ou en trans (transfert) [1].
La seconde voie, celle des modifications post-traductionnelles des histones comprend plusieurs types de modifications: acétylation, phosphorylation, ADP-ribosylation, méthylation ou encore ubiquitinylation. Elles affectent les extrémités amino- ou carboxy-terminales des quatre histones du coeur du nucléosome, situées en surface de celui-ci. Une combinaison de différentes modifications post-traductionnelles peut être associée spécifiquement à un état fonctionnel donné comme, par exemple, l’activation de la transcription. Ce véritable «langage» a conduit au concept de «code des histones» [2].
Enfin, la troisième voie, aujourd’hui l’objet d’un intérêt grandissant, met en jeu les variants d’histones. Ces variants sont présents chez tous les organismes eucaryotes et constituent des isoformes non alléliques des histones conventionnelles H2A, H2B, H3 et H4. Les variants, comme les histones classiques, sont constitués de trois hélices α reliées entre elles par deux courtes boucles L1 et L2. Cette structure est appelée histone fold et permet la dimérisation des histones (Figure 1B). Elle est conservée au cours de l’évolution. Au sein du nucléosome, la substitution d’une histone conventionnelle par un variant peut créer un nouveau complexe nucléo-protéique de structure et de stabilité modifiées. Cette modification pourrait favoriser le recrutement de facteurs régulateurs pour assurer les grandes fonctions nucléaires.
Deux classifications des variants d’histones ont été proposées. La première, basée sur la structure, distingue les variants homéomorphes des variants hétéromorphes. Les variants homéomorphes sont retrouvés dans toutes les classes d’histones, et ne diffèrent de leurs homologues conventionnels que par quelques substitutions d’acides aminés sans conséquence sur leur mobilité électrophorétique (à l’exception de H2A2) [3]. Au contraire, les variants hétéromorphes [4] présentent une extrémité carboxy-terminale dont la taille et la séquence en acides aminés sont suffisamment variables pour modifier de façon significative leur mobilité électrophorétique. Ces variants sont retrouvés dans les classes d’histones H2A et H2B et sont intégrés dans la chromatine dans des proportions variables. La seconde classification repose sur des critères plus fonctionnels: dépendance ou indépendance de la synthèse par rapport à la réplication, existence d’une spécificité tissulaire d’expression.
Dans cet article, nous avons choisi de nous focaliser sur trois variants majeurs de l’histone H2A associés à des fonctions spécifiques connues: les variants H2A.X, H2A.Z et macroH2A (Tableau I). Un quatrième variant, H2A-Bbd, a été récemment découvert, mais n’a fait l’objet que de rares études [5]. Quelle peut être l’origine de la spécificité fonctionnelle des variants ? L’histone H2A est la seule des quatre histones du coeur des nucléosomes dont l’extrémité carboxy-terminale s’étend au-delà de l’histone fold. Cette extrémité occupe une position critique puisqu’elle interagit avec l’ADN à son entrée et à sa sortie du nucléosome[6]. La comparaison de la séquence en acides aminés des trois variants de H2A indique clairement que, si les séquences correspondant à l’histone fold divergent faiblement, l’extrémité carboxy-terminale de chacun des variants est très variable (Figure3). Cette extrémité pourrait donc participer à la spécialisation fonctionnelle des variants de l’histone H2A.
Tableau I
Les variants de l’histone H2A chez l’homme.
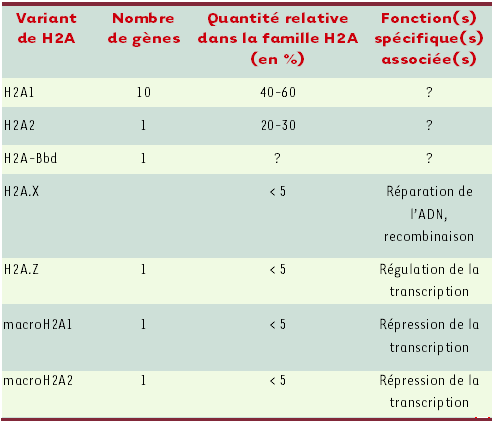
(d’après [51])
Figure 3
Comparaison des séquences des histones humaines de la famille H2A.
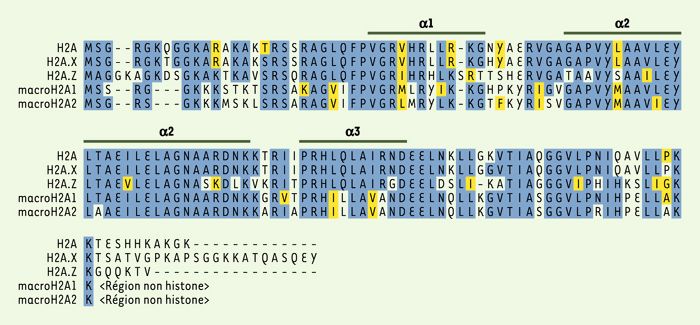
Les résidus identiques sont figurés en bleu et les résidus similaires en jaune. Les hélices α sont indiquées (d’après [52]). L’alignement a été réalisé avec le programme CLUSTALW.
L’histone H2A.X
Au début de la réplication, la quantité d’ARN codant pour les histones conventionnelles est augmentée de 10 à 50 fois. Cette augmentation est corrélée à une augmentation de la synthèse des protéines histones au cours de la phase S. Le gène du variant H2A.X code pour deux ARN. Le premier, de grande taille, est polyadénylé, et son expression est indépendante de la réplication [7]. Le second, environ trois fois plus court, est tronqué dans sa région 3’ non traduite (UTR), n’est pas polyadénylé et son expression est dépendante de la réplication: sa concentration décroît lorsque les cellules sont traitées avec des inhibiteurs de la réplication. De façon remarquable, la protéine H2A.X est retrouvée chez tous les eucaryotes supérieurs. Chez l’homme, elle est présente dans tous les types cellulaires testés. Elle subit plusieurs types de modifications post-traductionnelles: acétylation [8], ADP-ribosylation [9] et phosphorylation [10]. Cette dernière modification a été décrite comme directement associée à la présence du variant au niveau des sites de cassures de l’ADN. La forme phosphorylée du variant H2A.X est connue sous le nom de γH2A.X.
Un rôle dans la réparation des coupures double brin de l’ADN
L’histone γH2A.X semble impliquée dans le maintien de l’intégrité du génome en réponse aux lésions double brin de l’ADN. Elle est dans ce cas phosphorylée par la kinase ATM (ataxia telangiectasia mutated) [11] sur un motif conservé qui comprend la sérine 139. Des expériences d’immunofluorescence montrent que dans les noyaux des cellules soumises à des radiations ionisantes [12] ou en apoptose [16], H2A.X est rapidement phosphorylé. γH2A.X se distribue alors sous forme de foyers dans les noyaux interphasiques et sous forme de bandes sur les chromosomes métaphasiques, révélant ainsi le recrutement massif de ce variant dans les régions lésées. Parmi les protéines présentes au sein de ces foyers, on trouve également des protéines impliquées dans la réparation de l’ADN comme les protéines du complexe MRN (Mre11/Rad50/Nbs1), Rad51, BRCA1 ainsi que la protéine p53P1 (suppresseur de tumeur associé à la protéine p53) [13]. γH2A.X semble jouer un rôle clé dans l’initiation de la formation de ces complexes. De plus, le recrutement des facteurs de réparation au sein des foyers nucléaires à la suite de lésions de l’ADN est inhibé par la Wortmannine, inhibiteur fongique des kinases PI3KK responsables de la phosphorylation du variant [14]. L’implication de la forme phosphorylée du variant dans les mécanismes de recombinaison est également suggérée par la présence de foyers γH2A.X au niveau des sites de coupures double brin produits au cours de la méiose [15].
Si γH2A.X et les protéines associées au sein de ces foyers participent probablement au maintien de l’intégrité des chromosomes, les signaux physiologiques qui activent les systèmes de surveillance ne sont pas encore décrits. Au cours de l’apoptose, γH2A.X apparaît dès la fragmentation de l’ADN, et il semblerait que l’introduction dans la cellule d’une endonucléase à activité apoptotique suffise à induire la forme phosphorylée de ce variant [16].
Enfin, récemment, une accumulation nucléaire de γH2A.X a également été décrite dans les cellules T, au locus du récepteur α soumis au clivage V(D)J induit par la protéine activatrice de la recombinaison [17]. Une accumulation du variant phosphorylé a aussi été observée au niveau du locus TCR (T cell receptor) siège de la réaction de recombinaison permettant la fusion des gènes codant pour les régions variables et constantes des chaînes lourdes d’immunoglobulines. Cette réaction de recombinaison est altérée dans les souris H2A.X–/–, ce qui suggère que γH2A.X joue un rôle fonctionnel essentiel dans ce processus [18].
H2A.X et surveillance de la réplication
Parallèlement à son implication dans la reconnaissance et la réparation des coupures double brin de l’ADN, γH2A.X pourrait participer au contrôle de la réplication de l’ADN. Si on inhibe la réplication par de l’hydroxyurée ou par une faible irradiation UV, une accumulation du variant se produit dans les fourches de réplication dont la progression est arrêtée [19]. Dans ce cas, la formation de foyers γH2A.X est sous le contrôle de la kinase ATR (ataxia teleangiestasia and rad3 related protein) et non de ATM comme lors de la réponse aux radiations ionisantes. Ainsi, la cellule serait capable d’analyser la nature des dommages subis par l’ADN et d’activer en retour l’une ou l’autre des deux kinases.
H2A.X et structure chromatinienne
H2A.X, comme H2A.Z [20] et macroH2A [21], est présente dans les nucléosomes et un certain nombre de travaux suggèrent que la phosphorylation de H2A.X est associée à des modifications structurales de la chromatine. Au cours de la reconstitution in vitro de nucléosomes à partir d’extraits d’ovocytes de Xénope, une phosphorylation des variants d’histones est observée. Elle se produit pendant la phase précoce de la réaction d’assemblage, lors de l’association du variant H2A.X à l’ADN. Cette phosphorylation transitoire persiste quelques heures et semble importante pour structurer la fibre chromatinienne: une inhibition sélective de la phosphorylation de H2A.X perturbe l’espacement régulier des nucléosomes [22]. De même, l’incubation de noyaux de spermatozoïdes humains en présence d’ions magnésium et d’ATP conduit à un remodelage chromatinien corrélé à une phosphorylation des protamines et de l’histone H2A. En particulier, l’accroissement de l’espace entre deux nucléosomes s’accompagne, dans ce cas précis, de la phosphorylation du variant H2A.X [23].
Enfin, dernièrement, un travail réalisé chez la levure par Downs et al. [24] semble confirmer l’hypothèse selon laquelle H2A.X entraînerait des modifications structurales de la chromatine. Chez cet organisme, l’histone H2A.1 possède un motif de phosphorylation identique à celui de la région carboxy-terminale du variant H2A.X, absent de la levure. Ce site de phosphorylation est sous le contrôle, comme dans le cas de H2A.X, d’une kinase de la famille des PI3KK. La délétion de ce site de phosphorylation de H2A.1 entraîne une altération de la structure chromatinienne, avec en particulier une sensibilité accrue de la chromatine à la digestion par une endonucléase.
L’histone H2A.Z
Le gène codant pour H2A.Z[*] est retrouvé chez des organismes comme S. pombe [25], S. cerevisiae [26] et Tetrahymena [27], où sa transcription est réglée au cours du cycle cellulaire et partiellement dépendante de la réplication. En revanche, chez les mammifères, son expression est découplée de la synthèse de l’ADN [28].
Une fonction conservée et spécifique
La séquence peptidique du variant H2A.Z présente 60% d’homologie avec celle de l’histone H2A et 90% d’homologie inter-espèce. Jackson et Gorovsky [29] ont montré que chez la levure S. cerevisiae, le ralentissement de croissance associé à l’invalidation du gène codant pour H2A.Z peut être corrigé par l’introduction du gène du protozoaire cilié Tetrahymena thermophilia. En revanche, l’invalidation du gène codant pour H2A et la létalité associée ne sont pas corrigées par le gène codant pour H2A.Z. D’autres travaux chez la levure [30] ont montré que la fonction de H2A.Z est distincte de celle de H2A. En effet, au sein du nucléosome, l’interaction entre l’histone H4 et le dimère (H2A-H2B) met en jeu une tyrosine de l’histone H4. Une mutation de cette tyrosine induit un arrêt de la croissance des cellules à température restrictive, à tous les stades de division. La surexpression de H2A.Z restaure un phénotype normal chez ces mutants, alors que la surexpression d’autres protéines homologues de H2A (HTA1, BUR6) reste sans effet.
Un rôle essentiel dans la viabilité et le développement
Les levures S.pombe délétées du gène H2A.Z croissent lentement et présentent des défauts de ségrégation des chromosomes mitotiques. De même, le rôle essentiel du gène H2A.Z a été montré chez Tetrahymena [31], chez la drosophile [32] et chez la souris [33], organismes pour lesquels l’invalidation du gène est létale. Chez la souris, le croisement d’individus hétérozygotes (H2A.Z+/-), fertiles et normaux, ne produit aucun descendant homozygote. Pour confirmer le rôle essentiel de H2A.Z au cours du développement précoce murin, les auteurs ont utilisé la technique d’interférence de fonction génique par injection d’ARN double brin. Dans les oeufs fécondés de souris, l’injection d’ARN double brin H2A.Z n’empêche pas le développement jusqu’au stade blastocyste, mais inhibe l’attachement de l’embryon et la poursuite de sa croissance. Au vu de ces résultats, H2A.Z semble donc impliqué dans des fonctions spécifiques liées à la prolifération, la différenciation et/ou le développement.
H2A.Z et régulation de la transcription
Les données cristallographiques obtenues par Suto et al. [34] indiquent que la trajectoire de l’ADN qui s’enroule autour de l’octamère d’histones ayant incorporé H2A.Z ne serait pas modifiée. En revanche, les acides aminés spécifiques du variant H2A.Z déstabiliseraient l’association des dimères (H2A.Z-H2B) et (H3-H4). Cette déstabilisation pourrait faciliter l’accessibilité de la chromatine à des facteurs de transcription. Ces affirmations, bien que spéculatives, sont confortées par d’autres études in vitro (ultracentrifugation analytique sur nucléosomes reconstitués et sédimentation) [35]. Elles indiquent un effet déstabilisant de H2A.Z sur les interactions intra et inter-nucléosomales, déstabilisation nécessaire au recrutement de la machinerie transcriptionnelle.
Les premiers résultats d’immunolocalisation chez Tetrahymena ont montré que H2A.Z est associée à la chromatine transcriptionnellement active [36]. Dans les cellules végétatives de Tetrahymena, l’histone H2A.Z est détectée dans les macronoyaux transcriptionnellement actifs, et absente des micronoyaux inactifs [37]. De plus, au cours de la conjugaison, H2A.Z est exprimée dans les micronoyaux juste avant leur activation transcriptionnelle. Des approches biochimiques ont permis de mettre en évidence une interaction spécifique de l’extrémité carboxy-terminale de H2A.Z avec la grande sous-unité de l’ARN polymérase II. Par ailleurs, chez les levures délétées de H2A.Z, des expériences d’immunoprécipitation de la chromatine (ChlP) indiquent un défaut de recrutement de l’ARN polymérase et de la Tata-binding-protein sur le promoteur des gènes GAL 1 à 10 dans des conditions physiologiques pourtant favorables à l’activation de ces gènes [38]. Des levures délétées du gène codant pour H2A.Z ou pour SNF2, un autre gène impliqué dans le remodelage transcriptionnel de la chromatine, ne présentent pas de défauts sévères de croissance. En revanche, la croissance de doubles mutants de ces gènes est sévèrement altérée. Un résultat identique est obtenu pour des doubles mutants délétés des gènes codant pour H2A.Z et pour différentes sous-unités du complexe SAGA (Spt-Ada-Gcn5-acetyltransferase), complexe responsable de l’acétylation des histones. Ces résultats indiquent qu’en l’absence du variant H2A.Z, l’activité de complexes de remodelage ou de complexes modifiant post-traductionnellement les histones devient essentielle pour la viabilité. Ils plaident également pour un rôle redondant, au moins partiellement, des trois voies de modulation structurale du nucléosome que sont la mobilisation de complexe de remodelage, les modifications post-traductionnelles et l’incorporation de variants d’histones.
Si les travaux cités préalablement lient fonctionnellement H2A.Z à l’activation de la transcription, ce variant semble être également impliqué dans la répression transcriptionnelle. Chez la drosophile, Leach et al. [39] ont décrit la localisation de H2A.Z à la fois sur des chromosomes polyténiques et diploïdes. H2A.Z est distribuée sur l’ensemble de la chromatine, y compris en dehors des sites de transcription actifs. Ce patron de distribution n’est pas directement corrélé au degré de compaction de la chromatine dans le noyau, contrairement à ce que l’on observe pour l’histone H2A. De plus, des expériences d’immunoprécipitation de la chromatine montrent une association de H2A.Z aussi bien à des gènes transcrits qu’à des gènes non transcrits ou des séquences non codantes. Plus encore, les travaux de Dhillon et al. chez la levure démontrent un effet répressif probablement direct de H2A.Z sur la transcription au niveau de régions silencieuses (locus HMR) [40]. En conclusion, il semble que H2A.Z puisse jouer un rôle à la fois positif et négatif sur l’expression des gènes.
L’histone macroH2A
Ce variant doit son nom à sa taille inhabituelle (372acides aminés), qui représente près de trois fois la taille de l’histone H2A conventionnelle. Le premier tiers de la protéine possède environ 64% d’homologie de séquence avec l’histone H2A. Les deux tiers restant ne contiennent aucun motif de type histone fold [21]. Deux sous-types de macroH2A ont été identifiés: macroH2A1 et macroH2A2, codés chacun par un gène propre constitué de huit exons codants. Ces deux sous-types présentent 84% d’homologie au niveau de la séquence histone fold, et 68% dans la région non histone. Contrairement aux variants H2A.X et H2A.Z, il n’existe que peu de données génétiques ou biochimiques concernant macroH2A. MacroH2A1 est le sous-type le plus étudié.
Une fonction conservée au cours de l’évolution
MacroH2A1 est présent à la fois chez les oiseaux et les mammifères. Son gène code pour deux isoformes, 1.1 ou 1.2, issues d’un épissage alternatif, qui présentent chacune un motif de type «leucine zipper» retrouvé dans certains facteurs de transcription. Ces deux isoformes ne diffèrent l’une de l’autre que par une vingtaine d’acides aminés dans la région non histone. L’analyse de leur taux d’expression tissulaire indique des profils d’expression différents. La forme 1.1 est retrouvée à des taux variables dans les tissus adultes riches en cellules quiescentes. La forme 1.2 est retrouvée principalement dans les tissus à forte prolifération cellulaire (foie foetal, thymus adulte, testicule) [41]. Ces données suggèrent une fonctionnalité différente de ces sous-types. La séquence protéique, quelle que soit l’isoforme considérée, est remarquablement conservée puisque l’homologie est de 95% entre les classes oiseau et mammifère. Ce degré de conservation est très proche de l’homologie rencontrée pour l’histone H2A (98% oiseau/mammifère).
Un rôle dans l’inactivation du chromosome X, et au-delà…
En 1998, des résultats obtenus par Costanzi et Pehrson [42] semblaient démontrer que l’histone macroH2A, bien que présente à la fois dans les cellules mâles et femelles, était concentrée dans les noyaux de cellules femelles dans la région nucléaire occupée par le chromosome X inactif. Les auteurs proposaient un rôle possible de macroH2A dans les mécanismes moléculaires liés à l’inactivation du chromosome X.
L’inactivation du chromosome X chez les mammifères femelles se met en place très tôt au cours de l’embryogenèse et conduit à une inactivation quasi totale des gènes portés par ce chromosome [43, 44]. Les mécanismes moléculaires impliqués dans l’inactivation du chromosome X sont encore loin d’être élucidés. Ils mettent en jeu un transcrit non traduit, le transcrit Xist (X inactivation specific transcript) ((→) m/s 2000, nos 6-7, p.818) qui s’accumule en cis au niveau du chromosome X inactif [45] et dont le rôle n’a pas été élucidé. Aucun partenaire protéique de cet ARN et du chromosome X inactif n’ayant été jusqu’alors décrit, l’implication possible du variant d’histone macroH2A dans les mécanismes d’inactivation du chromosome X a reçu un large écho.
L’inactivation du chromosome X met en jeu 3 étapes: le déclenchement de l’inactivation, sa propagation le long du chromosome X et son maintien dans les cellules somatiques [46]. Dans les cellules embryonnaires ES, la redistribution du variant macroH2A au niveau du chromosome X inactif est postérieure aux étapes de déclenchement et de propagation de l’inactivation [47]. Dans le noyau de cellules ES en cours de différentiation, l’apparition tardive de l’association de macroH2A avec des gènes portés par le chromosome X a confirmé que macroH2A ne jouerait pas un rôle clef dans le processus de mise en place de l’inactivation [48]. Cependant, le rôle de macroH2A dans le maintien de l’inactivation n’a pas été non plus clairement établi. Il existe par exemple des lignées tumorales dans lesquelles il n’y a pas d’accumulation de macroH2A au niveau du chromosome X inactivé [47]. Diverses observations suggèrent que le rôle d’inactivation transcriptionnelle de ce variant n’est sans doute pas limité au chromosome X. En effet, d’un point de vue topographique, si la distribution de macroH2A dans les cellules femelles a plaidé initialement en faveur de son rôle dans l’inactivation du chromosome X, un marquage diffus de la protéine est également observé dans le reste du noyau. Ce type de marquage est aussi retrouvé dans les cellules mâles et la quantité de transcrits macroH2A1 est identique pour un tissu donné, dans les cellules mâles et femelles [49]. D’un point de vue structural, il a été montré que l’enrichissement du variant macroH2A sur le chromosome X inactif ne serait pas spécifique, puisque par exemple les histones conventionnelles du coeur du nucléosome (H2A, H2B, H3) s’accumulent aussi sur ce chromosome [50]. D’un point de vue fonctionnel, le ciblage, dans les cellules de mammifères, de la région non-histone du variant vers la région promotrice d’un gène rapporteur (par fusion au domaine de fixation à l’ADN du facteur de transcription Gal4) a révélé son rôle probable dans la répression de la transcription [50]. L’ensemble de ces résultats suggère une interprétation plus large du rôle de macroH2A que celui qui lui a été assigné jusqu’ici. Ce variant serait effectivement associé à la chromatine transcriptionnellement inactive, sans être exclusivement associé au chromosome X inactif.
Conclusions
L’ensemble des données présentées dans cette synthèse illustre la diversité des mécanismes auxquels les variants d’histones de coeur du nucléosome sont associés: surveillance de l’intégrité du génome, prolifération, différenciation... L’identification des facteurs protéiques associés aux variants d’histone et la compréhension des mécanismes de ciblages des variants dans des régions précises du génome sont l’enjeu des recherches à venir. Ces travaux sont indispensables pour déterminer le rôle fonctionnel des variants et comprendre la manière dont les acteurs impliqués dans les trois voies de modifications du nucléosome coopèrent. Cette connaissance sera essentielle pour identifier les dysfonctionnements des machineries chromatiniennes susceptibles d’être impliqués dans différentes pathologies. En particulier, la compréhension des anomalies structurales et fonctionnelles de la chromatine, observées dans un nombre croissant de tumeurs, pourrait être à l’origine de nouveaux développements diagnostiques et thérapeutiques.
Note ajoutée aux épreuves
Depuis la rédaction de cet article, plusieurs publications majeures ont encore éclairé le rôle de chacun des variants de l’histone H2A. H2A.X ne constituerait pas le signal initial de ciblage des complexes protéiques de réparation à proximité des zones d’ADN présentant des lésions mais participerait plutôt au maintien de ces protéines dans les régions chromatiniennes endommagées [53]. De même, un retard de croissance, une immunodéficience et une infertilité ont été décrits chez les souris mutantes H2A.X–/– [54]. En employant la technologie des puces à ADN, Meneghnini et al. ont démontré chez la levure l’activité «anti-répression» de H2A.Z: le variant protège des gènes proches de régions télomériques de la répression transcriptionnelle et de l’hétérochromatinisation imposées par des protéines de la famille SIR [55]. Enfin, Angelov et al. ont très récemment utilisé la reconstruction de nucléosomes in vitro pour mettre en évidence les propriétés des différents domaines de macroH2A [56]. Les résultats indiquent que la région histone de macroH2A s’oppose au remodelage des nucléosomes par le complexe SWI/SNF. La grande région non histone du variant interfère, elle, avec la fixation du facteur de transcription NF-κB.
Parties annexes
Remerciements
Les auteurs remercient Catherine Souchier et Stefan Dimitrov pour leur relecture critique de ce manuscrit. Pierre-Yves Perche a bénéficié d’une bourse de thèse de l’Association pour la Recherche sur le Cancer.
Note
-
[*]
Le variant H2A.Z est nommé différemment suivant les organismes où il est retrouvé (S. cerevisiae: HTA3 ou HTZ1, S. pombe: PHT1, T. Thermophila: hv1, D. melanogaster: H2AvD). Par souci de clarté, seul le nom H2A.Z est employé dans cette synthèse.
Références
- 1. Fry CJ, Peterson CL. Chromatin remodeling enzymes: who’s on first? Curr Biol 2001; 11: R185-97.
- 2. Strahl BD, Allis CD. The language of covalent histone modifications. Nature 2000; 403: 41-5.
- 3. Franklin SG, Zweidler A. Non-allelic variants of histones 2a, 2b and 3 in mammals. Nature 1977; 266: 273-5.
- 4. West MH, Bonner WM. Histone 2A, a heteromorphous family of eight protein species. Biochemistry 1980; 19: 3238-45.
- 5. Chadwick BP, Willard HF. A novel chromatin protein, distantly related to histone H2A, is largely excluded from the inactive X chromosome. J Cell Biol 2001; 152: 375-84.
- 6. Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 1997; 389: 251-60.
- 7. Mannironi C, Bonner WM, Hatch CL. H2A.X. a histone isoprotein with a conserved C-terminal sequence, is encoded by a novel mRNA with both DNA replication type and polyA 3’ processing signals. Nucleic Acids Res 1989; 17: 9113-26.
- 8. Pantazis P, Bonner WM. Quantitative determination of histone modification. H2A acetylation and phosphorylation. J Biol Chem 1981; 256: 4669-75.
- 9. Lindner H, Wesierska-Gadek J, Helliger W, Puschendorf B, Sauermann G. Identification of ADP-ribosylated histones by the combined use of high- performance liquid chromatography and electrophoresis. J Chromatogr 1989; 472: 243-9.
- 10. Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 1998; 273: 5858-68.
- 11. Burma S, Chen BP, MurphyM, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem 2001; 276: 42462-7.
- 12. Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol 1999; 146: 905-16.
- 13. Schultz LB, Chehab NH, Malikzay A, Halazonetis TD. p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J Cell Biol 2000; 151: 1381-90.
- 14. Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol 2000; 10: 886-95.
- 15. Mahadevaiah SK, Turner JM, Baudat F, et al. Recombinational DNA double-strand breaks in mice precede synapsis. Nat Genet 2001; 27: 271-6.
- 16. Rogakou EP, Nieves-Neira W, Boon C, Pommier Y, Bonner WM. Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J Biol Chem 2000; 275: 9390-5.
- 17. Chen HT, Bhandoola A, Difilippantonio MJ, et al. Response to RAG-mediated VDJ cleavage by NBS1 and gamma-H2AX. Science 2000; 290: 1962-5.
- 18. Petersen S, Casellas R, Reina-San-Martin B, et al. AID is required to initiate Nbs1/gamma-H2AX focus formation and mutations at sites of class switching. Nature 2001; 414: 660-5.
- 19. Ward IM, Chen J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem 2001; 276: 47759-62.
- 20. Hatch CL, Bonner WM, Moudrianakis EN. Minor histone 2A variants and ubiquinated forms in the native H2A:H2B dimer. Science 1983; 221: 468-70.
- 21. Pehrson JR, Fried VA. MacroH2A, a core histone containing a large nonhistone region. Science 1992; 257: 1398-400.
- 22. Kleinschmidt JA, Steinbeisser H. DNA-dependent phosphorylation of histone H2A.X during nucleosome assembly in Xenopus laevis oocytes: involvement of protein phosphorylation in nucleosome spacing. Embo J 1991; 10: 3043-50.
- 23. Banerjee S, Smallwood A, Hulten M. ATP-dependent reorganization of human sperm nuclear chromatin. J Cell Sci 1995; 108: 755-65.
- 24. Downs JA, Lowndes NF, Jackson SP. A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature 2000; 408: 1001-4.
- 25. Carr AM, Dorrington SM, Hindley J, Phear GA, Aves SJ, Nurse P. Analysis of a histone H2A variant from fission yeast: evidence for a role in chromosome stability. Mol Gen Genet 1994; 245: 628-35.
- 26. Spellman PT, Sherlock G, Zhang MQ, et al. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol Biol Cell 1998; 9: 3273-97.
- 27. White EM, Shapiro DL, Allis CD, Gorovsky MA. Sequence and properties of the message encoding Tetrahymena hv1, a highly evolutionarily conserved histone H2A variant that is associated with active genes. Nucleic Acids Res 1988; 16: 179-98.
- 28. Hatch CL, Bonner WM. The human histone H2A.Z gene. Sequence and regulation. J Biol Chem 1990; 265: 15211-8.
- 29. Jackson JD, Gorovsky MA. Histone H2A.Z has a conserved function that is distinct from that of the major H2A sequence variants. Nucleic Acids Res 2000; 28: 3811-6.
- 30. Santisteban MS, Kalashnikova T, Smith MM. Histone H2A.Z regulates transcription and is partially redundant with nucleosome remodeling complexes. Cell 2000; 103: 411-22.
- 31. Liu X, Bowen J, Gorovsky MA. Either of the major H2A genes but not an evolutionarily conserved H2A.F/Z variant of Tetrahymena thermophila can function as the sole H2A gene in the yeast Saccharomyces cerevisiae. Mol Cell Biol 1996; 16: 2878-87.
- 32. Clarkson MJ, Wells JR, Gibson F, Saint R, Tremethick DJ. Regions of variant histone His2AvD required for Drosophila development. Nature 1999; 399: 694-7.
- 33. Faast R, Thonglairoam V, Schulz TC, et al. Histone variant H2A.Z is required for early mammalian development. Curr Biol 2001; 11: 1183-7.
- 34. Suto RK, Clarkson MJ, Tremethick DJ, Luger K. Crystal structure of a nucleosome core particle containing the variant histone H2A.Z. Nat Struct Biol 2000; 7: 1121-4.
- 35. Abbott DW, Ivanova VS, Wang X, Bonner WM, Ausio J. Characterization of the stability and folding of H2A.Z chromatin particles: implications for transcriptional activation. J Biol Chem 2001; 276: 41945-9.
- 36. Stargell LA, Bowen J, Dadd CA, et al. Temporal and spatial association of histone H2A variant hv1 with transcriptionally competent chromatin during nuclear development in Tetrahymena thermophila. Genes Dev 1993; 7: 2641-51.
- 37. Allis CD, Richman R, Gorovsky MA, et al. hv1 is an evolutionarily conserved H2A variant that is preferentially associated with active genes. J Biol Chem 1986; 261: 1941-8.
- 38. Adam M, Robert F, Larochelle M, Gaudreau L. H2A.Z is required for global chromatin integrity and for recruitment of RNA polymerase II under specific conditions. Mol Cell Biol 2001; 21: 6270-9.
- 39. Leach TJ, Mazzeo M, Chotkowski HL, Madigan JP, Wotring MG, Glaser RL. Histone H2A.Z is widely but nonrandomly distributed in chromosomes of Drosophila melanogaster. J Biol Chem 2000; 275: 23267-72.
- 40. Dhillon N, Kamakaka RT. A histone variant, Htz1p, and a Sir1p-like protein, Esc2p, mediate silencing at HMR. Mol Cell 2000; 6: 769-80.
- 41. Pehrson JR, Costanzi C, Dharia C. Developmental and tissue expression patterns of histone macroH2A1 subtypes. J Cell Biochem 1997; 65: 107-13.
- 42. Costanzi C, Pehrson JR. Histone macroH2A1 is concentrated in the inactive X chromosome of female mammals. Nature 1998; 393: 599-601.
- 43. Heard E, Clerc P, Avner P. X-chromosome inactivation in mammals. Annu Rev Genet 1997; 31: 571-610.
- 44. Cohen DE, Lee JT. X-chromosome inactivation and the search for chromosome-wide silencers. Curr Opin Genet Dev 2002; 12: 219-24.
- 45. Clerc P. Tsix et Xist, antisens et sens: du verlan dans l’inactivation du chromosome X de la souris. Med Sci 2000; 16: 818-9.
- 46. Blanche J. Encore l’inactivation du chromosome X chez la souris. Med Sci 1998; 14: 976.
- 47. Mermoud JE, Costanzi C, Pehrson JR, Brockdorff N. Histone macroH2A1.2 relocates to the inactive X chromosome after initiation and propagation of X-inactivation. J Cell Biol 1999; 147: 1399-408.
- 48. Rasmussen TP, Mastrangelo MA, Eden A, Pehrson JR, Jaenisch R. Dynamic relocalization of histone MacroH2A1 from centrosomes to inactive X chromosomes during X inactivation. J Cell Biol 2000; 150: 1189-98.
- 49. Csankovszki G, Panning B, Bates B, Pehrson JR, Jaenisch R. Conditional deletion of Xist disrupts histone macroH2A localization but not maintenance of X inactivation. Nat Genet 1999; 22: 323-4.
- 50. Perche PY, Vourc’h C, Konecny L, et al. Higher concentrations of histone macroH2A in the Barr body are correlated with higher nucleosome density. Curr Biol 2000; 10: 1531-4.
- 51. Rogakou EP, Sekeri-Pataryas KE. Histone variants of H2A and H3 families are regulated during in vitro aging in the same manner as during differentiation. Exp Gerontol 1999; 34: 741-54.
- 52. Wolffe A. Chromatin, structure and function, 3e éd. New York: Academic Press, 1992.
- 53. Celeste A, Fernandez-Capetillo O, Kruhlak MJ, et al. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat Cell Biol 2003; 5: 675-9.
- 54. Celeste A, Petersen S, Romanienko PJ, et al. Genomic instability in mice lacking histone H2AX. Science 2002; 296: 922-7.
- 55. Meneghini MD, Wu M, Madhani HD. Conserved histone variant H2A.Z protects euchromatin from the ectopic spread of silent heterochromatin. Cell 2003; 112: 725-36.
- 56. Angelov D, Molla A, Perche PY, et al. The histone variant macroH2A interferes with transcription factor binding and SWI/SNF nucleosome remodeling. Mol Cell 2003; 11: 1033-41.
Liste des figures
Figure 1
Structure schématique de la particule coeur du nucléosome.
A. La double hélice d’ADN s’enroule autour de l’octamère d’histones. Les extrémités amino-terminales des histones ne sont pas représentées. B. Le repliement (histone fold) des hélices α et des boucles de connexion (L) des histones H2A et H2B permet leur dimérisation.
Figure 2
Les trois voies de modification du nucléosome.
A. Déplacement en cis ou en trans des nucléosomes par un complexe de remodelage. B. Modification post-traductionnelle des extrémités des histones. C. Substitution d’une histone conventionnelle par un variant.
Figure 3
Comparaison des séquences des histones humaines de la famille H2A.
Les résidus identiques sont figurés en bleu et les résidus similaires en jaune. Les hélices α sont indiquées (d’après [52]). L’alignement a été réalisé avec le programme CLUSTALW.
Liste des tableaux
Tableau I
Les variants de l’histone H2A chez l’homme.
(d’après [51])