Résumés
Résumé
Au moment de la placentation, l’embryon s’entoure d’une couche de trophoblaste où s’organisent l’arborisation des villosités et la circulation utéro-placentaire. L’analyse des modalités de l’arrivée du sang maternel a mis en évidence une hypoxie physiologique s’interrompant en fin de premier trimestre. De plus, les cultures d’explants villositaires ont montré le rôle de l’oxygène dans la prolifération du trophoblaste extravillositaire, l’invasion de la décidue et le remodelage des artères spiralées. L’oxygène apparaît maintenant comme un facteur clé dans le mécanisme de la placentation en raison du contrôle qu’il exerce sur la transcription de certains gènes. Ce contrôle s’effectue grâce à une molécule sensible à la pression partielle d’oxygène. On attribue à l’oxygène un rôle dans la genèse de plusieurs pathologies de la grossesse. Il est notamment mis en cause dans la séquence des événements conduisant à la pré-éclampsie. Des corrélations ont été mises en évidence entre l’environnement en oxygène et l’arborisation anormale des villosités, dans le retard de croissance intra-utérin, ou lors de situations comme l’anémie maternelle ou la gestation en altitude.
Summary
At the time of placentation, the conceptus surrounds itself with a trophoblastic layer where the villous tree develops and the uteroplacental circulation takes place. Analysis of the modalities of maternal blood entrance demonstrated a physiological hypoxia ending with the first trimester of pregnancy. Moreover, cultures of first trimester villous explants have shown the role of oxygen in extravillous cytotrophoblast proliferation, decidual invasion and spiral artery remodeling. Oxygen appears to be a key factor controlling the mechanism of placentation by regulating the transcription of several genes, such as VEGF (vascular endothelial growth factor), leptin, etc. These genes are turned on or off as a function of oxygen partial pressure via an oxygen sensor. Oxygen is now considered to be implicated in the development of several pathologies of pregnancy. It is involved at different steps in the cascade of events leading to preeclampsia. Positive correlations have been observed between oxygen partial pressure and abnormal development of the villous tree in intrauterine growth retardation, and in maternal anemia or pregnancy in altitude.
Corps de l’article
Le déroulement de l’implantation de l’embryon humain est connu depuis les années 1960 grâce aux collections d’embryons d’Hertig et Rock de l’Institut Carnegie à Washington (États-Unis) et de Hamilton et Boyd à Cambridge (Royaume-Uni). Des données histologiques anciennes suggéraient que la circulation intervilleuse placentaire s’établissait vers la 4e semaine d’aménorrhée (SA) [1]. Des observations récentes réalisées au premier trimestre de la grossesse ont apporté un nouvel éclairage sur cette question. En effet, pour Hustin et Schaaps [2] et Jaffe et Jauniaux [3], la circulation sanguine ne pouvait pas être détectée dans les espaces intervilleux avant 12 SA. Une réévaluation anatomique et physiologique a donc été nécessaire pour étayer ce nouveau concept.
Par ailleurs, des observations in vitro effectuées sur des cultures d’explants de villosités du premier trimestre, ont montré le rôle de l’invasion trophoblastique dans les remaniements circulatoires de l’utérus gravide [4]. Les facteurs intervenant dans cette invasion ont été analysés, en particulier la concentration en oxygène (O2) du milieu, qui a suscité de nombreuses questions: quels sont les rapports entre ces observations et les nouvelles données circulatoires? Comment la modification de l’environnement en O2 apportée par la circulation sanguine initialement absente dans l’espace intervilleux joue-t-elle un rôle dans la placentation?
Ces événements anatomiques et leurs conséquences physiologiques ont fait l’objet de plusieurs controverses [3, 5] qui paraissent résolues. Désormais, ils s’intègrent dans un schéma cohérent, avec les résultats obtenus en cultures d’explants de villosités. Ce schéma qui remet en cause les mécanismes classiques de la placentation, permet d’apporter d’ores et déjà de nouvelles explications physiopathogéniques à la genèse de plusieurs pathologies de la grossesse.
Implantation et placentation
Lors de l’implantation, le trophectoderme cellulaire bordant le blastocyste forme, au contact de l’épithélium utérin, un syncytiotrophoblaste qui permet l’infiltration du conceptus entier dans l’endomètre à 6 jours post-conception (JPC) [6] (Figure 1). Dans ce syncytiotrophoblaste, des lacunes se constituent à 8 JPC qui, en fusionnant, créent un espace intervilleux entre les trabécules syncytiales [7]. Le cytotrophoblaste dérivé du trophectoderme prolifère et s’engouffre dans le syncytiotrophoblaste des villosités primordiales pour former des colonnes cytotrophoblastiques [8] ((→) m/s 1999, n°11, p.1236). Ces colonnes de cytotrophoblaste sont envahies par des cellules du mésoderme extra-embryonnaire où apparaissent les premiers vaisseaux foetaux vers 18 JPC. Le mésoderme repousse les cellules cytotrophoblastiques à la base des villosités donnant naissance au cytotrophoblaste extravillositaire. Certaines villosités qui atteignent l’endomètre sont appelées villosités crampons, les autres sont dites flottantes. Le cytotrophoblaste continue à proliférer à la base des villosités crampons. Il enveloppe toute la surface du conceptus et obstrue les artères spiralées utérines avant 43-44 JPC. Cette prolifération du cytotrophoblaste donne lieu à une première vague d’invasion du tissu interstitiel de l’endomètre et des artères spiralées par voie périvasculaire ou endovasculaire. Une seconde vague d’invasion allant jusqu’à la partie interne du myomètre et de ses vaisseaux survient vers 16-18 SA [4].
Figure 1
Formation des villosités primordiales.

Lors de l’implantation, le trophectoderme cellulaire bordant le blastocyste forme au contact de l’épithélium utérin un syncytiotrophoblaste qui permet l’infiltration du conceptus entier dans l’endomètre à 6 jours post-conception (JPC). Dans ce syncytiotrophoblaste, des lacunes se constituent à 8 JPC et, en fusionnant, créent un espace intervilleux entre les trabécules syncytiales. Le cytotrophoblaste dérivé du trophectoderme prolifère et s’engouffre dans le syncytiotrophoblaste des villosités primordiales (12 JPC) pour former des colonnes cytotrophoblastiques (CC). Ces colonnes de cytotrophoblaste sont envahies par des cellules du mésoderme extra-embryonnaire (14 JPC) où apparaissent les premiers vaisseaux foetaux vers 18 JPC. Le mésoderme repousse les cellules cytotrophoblastiques à la base des villosités donnant naissance au cytotrophoblaste extravillositaire (CEV). Certaines villosités qui atteignent l’endomètre sont appelées villosités crampons (VC), les autres sont dites flottantes (VF) et elles présentent parfois des îlots cytotrophoblastiques (IC).
Circulation précoce intervilleuse
Après décidualisation, l’endomètre est en mesure d’accueillir le conceptus dès l’ouverture de la fenêtre d’implantation. Les sécrétions des glandes utérines nourrissent le blastocyste avant et pendant l’implantation. Cette période histiotrophe est suivie d’une période plasmatrophe jusqu’à 12 semaines d’aménorrhée (SA), pendant laquelle le plasma traverse les bouchons cytotrophoblastiques filtrants des artères spiralées, pénètre dans l’espace intervilleux et baigne les villosités primordiales. En effet, les artères spiralées ne communiquent pas directement avec l’espace intervilleux [7]. Le réseau de sinusoïdes endométriaux qui enveloppe le conceptus joue probablement le rôle de shunt sanguin artério-veineux (Figure 2). Les communications directes entre les artères spiralées et l’espace intervilleux ne sont décelables qu’à 11-12 SA [9]. À partir de la 12e SA, la disparition des bouchons filtrants permet l’arrivée du sang dans l’espace intervilleux. Pour Hustin et Schaaps [2], l’absence de circulation sanguine dans l’espace intervilleux jusqu’à 12 SA était argumentée par des observations en hystéroscopie, en ultrasonographie et par l’examen de trois pièces d’hystérectomies gravides à 9-10 SA. Ces observations confirmaient des données histologiques anciennes [7]. D’autres observations avaient suggéré une arrivée plus précoce du sang (12-16 JPC) dans l’espace intervilleux [1], lui attribuant un rôle moteur dans le développement placentaire. Ce nouveau concept a suscité une première controverse à propos de la sensibilité de détection des flux sanguins faibles en échographie Doppler classique. Une seconde controverse s’est développée concernant la date d’ouverture des artères spiralées [3, 5]. L’échographie Doppler couleur montre actuellement que des flux sanguins intervilleux de type veineux sont observables à 5 SA et que des flux artériels apparaissent progressivement entre 8 et 12 SA [10, 11]. Ce laps de temps correspond à la période d’ouverture progressive des artères spiralées. On notera par ailleurs que la circulation ombilico-placentaire commence a être détectée en échographie Doppler couleur entre 5 et 7 SA et qu’elle l’est constamment à partir de 8 SA.
Figure 2
Circulation intervilleuse au premier trimestre.

À 8 semaines d’aménorrhée, les artères spiralées de la décidue sont partiellement bouchées par le cytotrophoblaste extravillositaire (CEV). Le plasma et quelques hématies maternelles circulent dans l’espace intervilleux (flèches jaunes). Le sang est court-circuité vers un réseau de sinusoïdes déciduaux et regagne les veines. À 12 semaines d’aménorrhée, les bouchons trophoblastiques ont disparu et le sang des artères utéro-placentaires s’écoule dans l’espace intervilleux. Le rôle des sinusoïdes déciduaux est devenu insignifiant. pO2: pression partielle d’oxygène.
Invasion trophoblastique et remodelage des artères spiralées
La paroi musculo-élastique des artères spiralées est responsable de leur forte résistance à l’écoulement du sang. Cette paroi est remodelée en raison de l’invasion par le cytotrophoblaste extravillositaire provenant de la base des villosités crampons. Il dissocie la paroi musculo-élastique des artères et se substitue par endroits à l’endothélium. Ces remaniements aboutissent à la formation d’artères utéro-placentaires [4] de faible impédance, et insensibles aux stimulations vasomotrices. Ils rendent possible l’augmentation du débit sanguin en fonction des besoins nutritionnels croissants de l’embryon et l’irrigation continue de l’espace intervilleux.
Au cours de l’invasion de la décidue et de ses vaisseaux, le cytotrophoblaste acquiert des phénotypes variés qui se traduisent par l’expression de marqueurs cytologiques spécifiques (Figure 3). Différentes sous-unités d’intégrines sont observées [12] selon que le cytotrophoblaste est de type épithélial (α3β4, α6β4), interstitiel (α1β3, α5β1) ou endovasculaire (α1β3, αvβ3). Leur expression est en rapport avec l’adhérence des cellules aux constituants de la matrice extracellulaire (α6: laminine, α5: fibronectine, α1: collagène). Le registre des sous-unités d’intégrines exprimées a été bien caractérisé dans les grossesses normales et, avec d’autres marqueurs, il permet d’évaluer la capacité de prolifération et d’invasion du cytotrophoblaste dans les grossesses pathologiques. Le cytotrophoblaste met également en oeuvre des métalloprotéases matricielles (MMP) synthétisées et activées au cours de la progression des cellules dans la matrice extracellulaire comme la gélatinase B (MMP9) qui remplace la gélatinase A (MMP2) [13].
Figure 3
Invasion trophoblastique (8e semaine d’aménorrhée).

La paroi musculo-élastique des artères spiralées est remodelée du fait de l’invasion par le cytotrophoblaste extravillositaire, en provenance de la base des villosités crampons. Celui-ci dissocie la paroi musculo-élastique des artères et se substitue par endroits à l’endothélium. Au cours de l’invasion de la décidue et de ses vaisseaux, le cytotrophoblaste acquiert des phénotypes variés qui se traduisent par l’expression de marqueurs cytologiques spécifiques. Différentes sous-unités d’intégrines sont observées selon que le cytotrophoblaste est de type épithélial (α3β4, α6β4), interstitiel (α1β3, α5β1) ou endovasculaire (α1β3, αVβ3). Le cytotrophoblaste met également en oeuvre des métalloprotéases matricielles (MMP) synthétisées et activées au cours de la progression des cellules dans la matrice extracellulaire comme la gélatinase B (MMP9) qui remplace la gélatinase A (MMP2). MIB-1: anticorps détectant KI67 en phase G1, S, G2 et M du cycle cellulaire ; TGFβ3: transforming growth factor β3; cadhérines E et V-E: molécules d’adhérence; HIF1α: hypoxia inducible factor-1α; MMP1, 2, 9: matrix metalloproteinase 1, 2, 9; PECAM-1: platelet-endothelial cell adhesion molecule-1.
Pression partielle d’oxygène dans l’espace intervilleux
Les mesures de la pression partielle d’O2 (pO2) par une méthode polarographique à 8-10 SA indiquent une valeur de 18 mmHg dans l’espace intervilleux [14] et de 40 mmHg dans les tissus déciduaux (Figure 2). Des valeurs similaires ont été obtenues par des mesures à l’électrode de Clark [15]. Avant 12 SA, du plasma pauvre en hématies circule dans l’espace intervilleux. Sa faible capacité de transport de l’O2 est responsable d’une pO2 basse qui peut déclencher la transcription cellulaire des gènes réglés par l’O2. À partir de 12 SA, le sang circule dans l’espace intervilleux, sa pO2 s’élève à 61 mmHg et celle des tissus déciduaux à 47 mmHg. Cette élévation est probablement en rapport avec la présence d’hémoglobine ou avec l’augmentation du débit sanguin. L’ouverture des artères utéro-placentaires, en augmentant la pO2 dans l’espace intervilleux, intervient dans la transcription des gènes réglés par l’hypoxie.
Environnement en O2, trophoblastes extravillositaire et villositaire
Des explants de villosités crampons ont été cultivés en présence de concentrations variables d’O2 afin d’évaluer le rôle de ce facteur dans la prolifération du cytotrophoblaste extravillositaire. Les explants de 8 SA, cultivés en présence de 2 % d’O2, développent des excroissances produites par la prolifération du cytotrophoblaste [16]. En présence de 20 % d’O2, ces excroissances n’apparaissent pas [17]. La prolifération cellulaire cesse, comme le confirment la diminution du taux de la cycline B et l’augmentation du taux de la protéine p21, deux molécules intervenant dans le cycle cellulaire.
L’ARNm du facteur inductible par l’hypoxie α (HIF1α) et l’ARNm du facteur de croissance transformant β3 (TGFβ3) sont significativement exprimés dans le cytotrophoblaste durant la prolifération des villosités crampons avant 11 SA, quand la pO2 intervilleuse est basse. En présence de 3 % d’O2, les explants de villosités de 5-8 SA forment des excroissances, synthétisent de la fibronectine et expriment la sous-unité α5 des intégrines et une activité MMP2 [18]. La prolifération est suivie d’une différenciation partielle sans acquisition du phénotype d’invasion (Figure 3). L’utilisation d’oligonucléotides antisens complémentaires de l’ARNm de l’HIF1α inhibe l’expression de TGFβ3 et induit celle de la sous-unité α1 des intégrines. En même temps, l’activité de la MMP9 apparaît. Ces modifications montrent l’acquisition d’un phénotype d’invasion induit par la baisse d’HIF1α et de TGFβ3. Les explants cultivés en présence de concentrations plus élevées d’O2 (jusqu’à 20 %) présentent les mêmes modifications. Entre 10-12 SA in vivo, lorsque la pO2 intervilleuse augmente, les ARNm d’HIF1α et de TGFβ3 diminuent également dans les villosités. De plus, les anticorps monoclonaux ou les oligonucléotides antisens dirigés contre l’endogline - protéine endothéliale associée au récepteur du TGFβ -stimulent la production d’excroissances et la synthèse de fibronectine dans les explants de villosités de 5-8 SA [19]. Ces effets sont inhibés par le TGFβ2, mais non par le TGFβ1 ou le TGFβ3. Le TGFβ2 serait donc impliqué dans la perte de polarité des cellules quittant la zone épithéliale de prolifération pour adopter le phénotype conjonctif. La prolifération et la différenciation du cytotrophoblaste extravillositaire en fonction de l’environnement en O2in vivo sont présentées dans la Figure 3.
Le trophoblaste des villosités flottantes assure la majeure partie des fonctions endocrine et de transport du placenta. En proliférant, le cytotrophoblaste permet le bourgeonnement de villosités nouvelles et leur arborisation. Par fusion cellulaire, il apporte ARNm, protéines, organites et noyaux au syncytiotrophoblaste dont l’activité transcriptionnelle est minimale [20]. Le syncytiotrophoblaste est expulsé dans la circulation maternelle après apoptose ou nécrose. Ces processus sont modifiés dans les pathologies impliquant l’environnement en O2. L’O2 peut intervenir à différentes étapes du renouvellement trophoblastique et altérer, en conséquence, les fonctions placentaires.
Gènes réglés par l’O2
Plusieurs facteurs de transcription inductibles par l’hypoxie, HIF 1, 2 et 3, ont été mis en évidence [21] ((→) m/s 2002, n°1, p.70). Dans le cas d’HIF1, la sous-unité HIF1α est stable en hypoxie et, après translocation dans le noyau, forme un hétérodimère avec la sous-unité HIF1β [22]. La liaison du dimère HIF1 à une séquence régulatrice HRE (consensus 5’-RCGTG-3’) de l’ADN avec d’autres co-facteurs induit la transcription de gènes contrôlés par l’O2. En normoxie, la prolyl-hydroxylase activée hydroxyle la proline 564 du domaine carboxy-terminal de l’HIF1α. Celui-ci fixe alors le facteur suppresseur de tumeur de von Hippel-Lindau (pVHL), qui, associé notamment aux élongines B et C et à la culline 2, forme un complexe ayant une activité E3-ubiquitine-ligase. L’HIF1α peut alors se lier à des ubiquitines et est dégradé par le protéasome. La prolyl-hydroxylase, en particulier l’isoforme PHD2, est donc un des capteurs potentiels d’O2 sensibles aux variations de pO2, mais d’autres molécules présentant un groupement hémique pourraient jouer ce rôle. La Figure 4 résume le mécanisme d’action d’HIF sur les gènes réglés par l’O2 dans le trophoblaste.
Figure 4
Pression partielle d’oxygène et expression des gènes dans le trophoblaste.
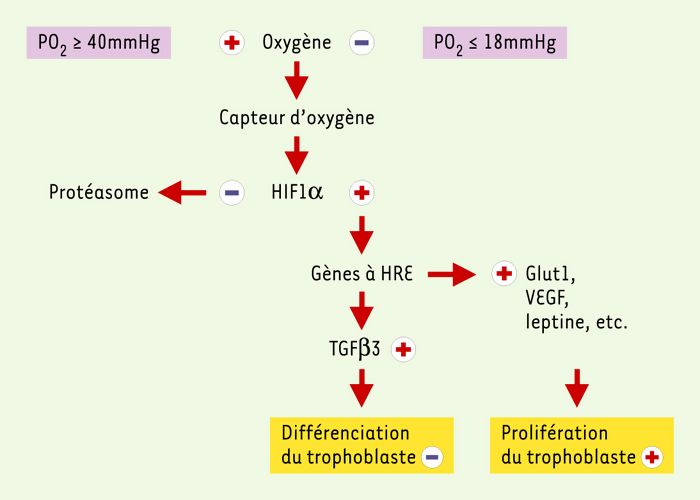
Plusieurs facteurs de transcription inductibles par l’hypoxie, HIF 1, 2 et 3 ont été mis en évidence. Dans le cas d’HIF1, la sous-unité HIF1α est stable en hypoxie et, après translocation dans le noyau, forme un hétérodimère avec la sous-unité HIF1. La liaison du dimère HIF1 à une séquence régulatrice HRE (consensus 5’-RCGTG-3’) de l’ADN avec d’autres co-facteurs induit la transcription de gènes contrôlés par l’O2: VEGF (vascular endothelial growth factor), Glut1 (glucose transporter 1), leptine, etc. L’activation des gènes à séquence HRE induit directement ou indirectement la prolifération trophoblastique. Certains membres de la famille du TGFβ comme TGFβ2, présentent une séquence d’ADN HRE fixant HIF1α. TGFβ3 pourrait présenter cette séquence et son effet s’exercerait en inhibant la différenciation. En normoxie, la prolyl-hydroxylase activée hydroxyle la proline 564 du domaine carboxy-terminal de l’HIF1α. Celui-ci fixe alors le facteur suppresseur de tumeur de von Hippel-Lindau, qui associé à d’autres éléments, forme un complexe ayant une activité E3 ubiquitine ligase. L’HIF1α peut alors se lier à des ubiquitines et il est dégradé par le protéasome. D’autres capteurs d’O2 à groupement hémique agissant sur HIFα ont été proposés.
L’invalidation du gène HIF1α chez la souris entraîne la mort des embryons à 10,5 jours de gestation, à la suite de malformations cardiaques et vasculaires similaires à celles observées après l’invalidation du facteur de croissance vasculaire (VEGF, vascular endothelial growth factor), mais cependant indépendantes. Les gènes sensibles à l’hypoxie répertoriés à ce jour [21] sont très variés: érythropoïétine, VEGF et certaines enzymes du métabolisme. Leur transcription dans les tissus normaux et tumoraux affecte l’hémodynamique, l’angiogenèse et le métabolisme. Une régulation de l’expression du VEGF par l’hypoxie a été observée chez le foetus de brebis au cours d’anémies chroniques induites: dans le tissu ventriculaire cardiaque, le taux d’ARNm de VEGF et celui de la protéine HIF1α sont multipliés par trois. Dans les cellules de choriocarcinome placentaire (lignées BeWo ou JEG), l’hypoxie stimule la production de VEGF [23]. La liste des gènes sensibles au facteur HIF1α s’allonge et inclut maintenant l’hème-oxygénase, l’endothéline 1, le transporteur de glucose Glut1 et la leptine [24] ((→) m/s 2001, n°4, p.496).
Grossesses pathologiques
Un certain nombre d’avortements précoces pourrait impliquer l’oxygène. En soumettant le trophoblaste à une pO2 élevée, l’ouverture précoce de l’espace intervilleux au sang maternel provoquerait une dégradation anticipée du facteur HIF1α et affecterait la prolifération du trophoblaste. Des signaux échographiques Doppler de type artériel anormalement précoces ont été détectés dans les espaces intervilleux de grossesses interrompues [25] ((→) m/s 2000, n°3, p.329).
La pré-éclampsie est une maladie affectant l’unité foeto-placentaire et la mère et qui se révèle le plus fréquemment au dernier trimestre. Généralement associée à une hypertension et à une protéinurie, la pré-éclampsie reste une maladie sévère en 2003. Elle touche environ 5 % des grossesses, provoque chaque année des décès maternels, est responsable d’un nombre élevé de césariennes et d’une morbidité néonatale importante. Le trophoblaste est impliqué au premier chef car l’invasion de l’endomètre, du myomètre et de leurs vaisseaux respectifs reste superficielle [26]. Les vaisseaux utérins présentent des lésions, comme l’accumulation de matériel fibrinoïde, la prolifération des myocytes et la thrombose des vaisseaux qui constituent les signes cliniques principaux de l’ischémie. La perfusion de l’espace intervilleux est réduite, mais cette réduction n’est pas spécifique de cette maladie [11]. Elle est associée à une augmentation de l’index de résistance obtenu par l’examen en échographie Doppler des artères utérines [27]. Les modifications circulatoires importantes observées dans la pré-éclampsie sévère suggèrent aussi une exagération générale du processus inflammatoire inhérent à la grossesse [28]. La complexité de cette maladie dans le placenta même et sa diffusion à des sites maternels divers transparaît dans l’hétérogénéité de ses manifestations cliniques. C’est pourquoi ses causes et ses conséquences ont souvent été confondues.
La cause initiale de la pré-éclampsie n’est pas clairement identifiée, mais la séquence des événements qui la déclenchent commence à être connue (Figure 5). Les artères spiralées conserveraient leur assise musculo-élastique dans leurs segments myométrial et parfois endométrial entraînant une perfusion inadéquate de l’espace intervilleux au premier trimestre. En effet, le cytotrophoblaste extravillositaire exprime uniquement des sous-unités d’intégrines épithéliales (α3β4, α6β4) et interstitielles (α5β1) [29] et n’acquiert pas le phénotype invasif. La MMP9, une protéase facilitant l’invasion de la décidue, est faiblement exprimée ainsi que les antigènes d’histocompatibilité de type G (HLA-G) indiquant que le trophoblaste ne se différencie pas normalement. Ces observations sont confirmées par les études in vitro qui montrent que le trophoblaste prolifère, mais n’entre pas en phase invasive. Une pO2 basse est probablement maintenue et l’HIF1α n’est pas dégradé. La concentration élevée de l’ARNm HIF2α placentaire a d’ailleurs été mise en cause récemment dans la pré-éclampsie. La persistance de HIF1α ou 2α expliquerait l’invasion réduite des artères spiralées (Figure 4).
Figure 5
Physiopathogénie possible de la pré-éclampsie.

La cause initiale de la pré-éclampsie n’est pas clairement identifiée mais la séquence des événements qui la déclenchent commence à être connue. Du fait d’une invasion trophoblastique incomplète, les artères spiralées conserveraient leur assise musculo-élastique dans leur segment myométrial et parfois endométrial, entraînant une perfusion inadéquate de l’espace intervilleux au premier trimestre. Le remodelage défectueux des artères spiralées provoque ensuite des altérations récurrentes de la circulation utéro-placentaire dues à la production d’ion superoxyde (O.2) au cours du stress oxydatif. Le dysfonctionnement de l’endothélium déclenche aussi une production accrue d’agents vasoconstricteurs (thromboxane et endothéline). L’expression de certaines cytokines pro-inflammatoires et immunorégulatrices produites au cours du stress oxydatif, comme le TNFα (tumor necrosis factor α) et les interleukines IL-1α et IL-10, est augmentée dans les placentas pré-éclamptiques et les concentrations de TNFα et d’IL-6 s’élèvent dans le sang maternel. On observe aussi dans le sang maternel un dérèglement du système de coagulation sanguine favorisant l’agrégation plaquettaire. Le mécanisme de diffusion des altérations placentaires à la mère reste obscur. L’excès de microfragments nécrotiques ou apoptotiques de syncytiotrophoblaste dans la circulation maternelle pourrait conduire à un dysfonctionnement généralisé de l’endothélium des vaisseaux maternels et participer ainsi à l’amplification de la réaction inflammatoire prédisposant à l’hypertension. RCIU: retard de croissance intra-utérin.
Le remodelage défectueux des artères spiralées entraîne ensuite des altérations récurrentes de la circulation utéro-placentaire dues à la production d’ion superoxyde (O.2) au cours du stress oxydatif. L’O.2 est normalement transformé par la superoxyde dismutase de type cytosolique en peroxyde d’hydrogène, également actif. L’invalidation du gène de la forme cytosolique de cette enzyme chez la souris est létale, en raison notamment de l’absence de développement placentaire; sa surexpression dans le trophoblaste in vitro ou dans la trisomie 21 est associée à la réduction de la formation du syncytiotrophoblaste [30]. Dans les modèles animaux d’hypertension, on observe une augmentation de la production d’O.2 mais aussi du monoxyde d’azote (NO). Cependant, la biodisponiblité du NO est diminuée, car il passe rapidement à l’état de complexe. Dans le placenta pré-éclamptique, bien que l’activité des NO synthases constitutive et inductible soit diminuée [31], l’augmentation des nitrotyrosines des protéines et des lipides peroxydés indique l’existence d’un stress oxydatif. L’O.2 associé au NO forme un peroxynitrite (ONOO-) toxique. Ce dernier inhibe la chaîne de transport des électrons, réduit les pools de NAD+ et d’ATP intracellulaires, déclenche la peroxydation des lipides et produit des cassures de l’ADN. Ces effets entraînent une sénescence prématurée des cellules endothéliales et trophoblastiques.
Certains dérivés oxygénés ont une action vasomotrice: l’O.2 a un effet vasoconstricteur et l’ONOO- atténue la réponse vasculaire aux agents constricteurs ou dilatateurs. Le dysfonctionnement de l’endothélium déclenche aussi une production accrue d’agents vasoconstricteurs (thromboxane et endothéline), une plus grande sensibilité vasculaire à l’angiotensine II, alors que la production d’agents relaxants (prostacycline et NO) est diminuée [32]. Ces modifications du tonus vasculaire provoquent une augmentation de l’impédance utérine en échographie Doppler (Figure 5).
L’expression de certaines cytokines inflammatoires et immunorégulatrices produites au cours du stress oxydatif, comme le TNFα (tumor necrosis factor α) et les interleukines IL-1α et IL-10, est augmentée dans les placentas pré-éclamptiques, et les concentrations de TNFα et d’IL-6 s’élèvent dans le sang maternel. On observe aussi dans le sang maternel un dérèglement du système de coagulation sanguine favorisant l’agrégation plaquettaire. Une libération accrue de molécules d’adhérence de type endothélial a été rapportée. L’endothélium de la circulation périphérique maternelle est en cause mais le placenta reste un acteur essentiel du déclenchement de la pré-éclampsie, puisque, dans la majorité des cas, l’hypertension et les autres manifestations cliniques disparaissent après la délivrance. Le mécanisme de diffusion des altérations placentaires à la mère reste obscur. L’excès de microfragments nécrotiques ou apoptotiques de syncytiotrophoblaste dans la circulation maternelle [33] pourrait conduire à un dysfonctionnement de l’endothélium des vaisseaux maternels et participer ainsi à l’amplification de la réaction inflammatoire maternelle prédisposant à l’hypertension. Au travers du trophoblaste villositaire prématurément sénescent, les échanges materno-foetaux s’appauvrissent, menant au retard de croissance intra-utérin qui peut s’accompagner d’une augmentation de l’impédance ombilico-placentaire.
L’oxygène peut aussi affecter l’ontogenèse des villosités foetales dont les phénotypes diffèrent selon les maladies et leur degré de gravité (Figure 6). Un aspect hypoxique (hyper-ramification et hyper-capillarisation des villosités terminales) a été noté dans la pré-éclampsie [34]. La situation d’hypoxie précoce maintenue dans la pré-éclampsie peut donc être suivie d’une ontogenèse villositaire compensatrice permettant une circulation normale du sang dans les vaisseaux villositaires. Un aspect hyperoxique (ramification villositaire déficiente associée à une angiogenèse) prédomine dans la pré-éclampsie avec retard de croissance intra-utérin associée à une vélocité diastolique absente ou inversée (VDAI) [35] ou dans le retard de croissance intra-utérin sans pré-éclampsie avec VDAI [34]. Une évaluation morphométrique des placentas pré-éclamptiques avec retard de croissance intra-utérin conclut aussi à une réduction du développement villositaire (longueur et surface) avec vasodilatation capillaire [36]. L’ischémie et l’hypoxie peuvent donc provoquer une altération profonde de l’ontogenèse se manifestant par la diminution du nombre des villosités terminales et des dilatations capillaires. Les capillaires présentent quelquefois une embolisation circulatoire érythrocytaire dans le retard de croissance intra-utérin avec VDAI [36]. Ces modifications peuvent être responsables de l’impédance élevée des vaisseaux ombilico-placentaires constatée dans le retard de croissance intra-utérin et la pré-éclampsie avec VDAI.
Figure 6
Environnement en oxygène et maladies.

L’oxygène peut aussi affecter l’ontogenèse des villosités foetales dont les phénotypes diffèrent selon les maladies et leur degré de gravité. Un aspect hypoxique (hyper-ramification et hyper-capillarisation des villosités terminales) a été noté dans la pré-éclampsie. Un aspect hyperoxique (ramification villositaire déficiente associée à une angiogenèse) prédomine dans la pré-éclampsie avec retard de croissance intra-utérin (RCIU) associée à une vélocité diastolique absente ou inversée (VDAI) ou dans le RCIU sans pré-éclampsie avec VDAI. L’anémie maternelle et la grossesse en altitude sont à l’origine de deux situations où l’appauvrissement en O2 a des répercussions caractéristiques sur la morphogenèse et l’angiogenèse villositaires. Dans les grossesses en altitude, on observe une ramification très importante des villosités terminales et une hyper-capillarisation.
L’anémie maternelle et la grossesse en altitude sont à l’origine de deux situations où l’appauvrissement en O2 a des répercussions caractéristiques sur la morphogenèse et l’angiogenèse villositaire. Dans les grossesses en altitude, on observe une ramification très importante des villosités terminales et une hyper-capillarisation [37]. Des mécanismes complexes ont été décrits dans l’hypoxie et l’hyperoxie, à propos de la synthèse et des mécanismes d’action du VEGF et du facteur de croissance placentaire (PlGF, placenta growth factor) [38]. Leur expression varie avec l’environnement en O2 et au cours de la gestation: l’expression du VEGF est suivie de celle du PlGF. Leurs effets sur le trophoblaste et l’arborisation des villosités (élongation des villosités, ramification et bourgeonnement terminal) ont été analysés antérieurement [39]. Le nombre des facteurs angiogéniques connus augmente ainsi que celui de leurs récepteurs dont certains sont solubles (sFlt-1), ce qui empêche pour l’instant d’envisager une interprétation cohérente des mécanismes d’ontogenèse des villosités, de vasculogenèse et d’angiogenèse. Cependant, l’analyse des concentrations sériques maternelles de sFlt-1 ou encore de leptine, qui paraissent plus élevées dans la pré-éclampsie, pourrait aider à la détection précoce de cette pathologie.
Conclusions
Les bouchons trophoblastiques assurant l’obstruction partielle de l’espace intervilleux jusqu’à 12 SA ont certainement une importance capitale dans la placentation. Le plasma maternel qui traverse les bouchons trophoblastiques perfuse l’espace intervilleux à faible pO2. Cette hypoxie physiologique en agissant sur des capteurs d’O2 déclenche la transcription de gènes sensibles à l’hypoxie, certains d’entre eux provoquant la prolifération du cytotrophoblaste extravillositaire. Après 12 SA, la pO2 plus élevée, observée à proximité des sinusoïdes endométriaux et dans l’espace intervilleux, diminue la transcription de ces gènes et provoque l’invasion de la décidue et la différenciation du trophoblaste. Les altérations de l’environnement en O2, en particulier l’hyperoxie, ont des effets délétères sur la placentation car l’expression et l’activité des enzymes anti-oxydants (superoxyde dismutase, catalase et glutathion peroxydase) apparaissent vers 8-9 SA [15]. L’O2 intervient non seulement dans la placentation précoce, mais aussi dans la morphogenèse et l’angiogenèse villositaires. Sa raréfaction paraît déclencher un processus morphogénétique compensateur dans la grossesse en altitude ou en cas d’anémie maternelle. Une compensation similaire peut être observée dans certains cas de pré-éclampsie alors que, dans d’autres cas, l’altération du développement villositaire due au stress oxydatif s’amplifie. Le stress oxydatif, l’intervention des cytokines dans les processus inflammatoire et immunorégulateur ainsi que l’incidence de l’apoptose et de la nécrose devront être mieux analysés pour comprendre l’étiologie de cette maladie complexe qui met en danger la vie de la mère et celle de l’enfant. À bien des égards, par son rapport particulier à l’oxygène, le placenta se singularise des autres organes [40].

Parties annexes
Remerciements
Les auteurs remercient Lionel Carbillon et Christian Vervelle pour leur aide constructive dans la rédaction de cet article.
Références
- 1. Hertig AT, Rock J, Adams EC. A description of 34 human ova within the first 17 days of development. Am J Anat 1956; 98: 435-93.
- 2. Hustin J, Schaaps JP. Echographic and anatomic studies of the maternotrophoblastic border during the first trimester of pregnancy. Am J Obstet Gynecol 1987; 157: 162-8.
- 3. Jaffe R, Jauniaux E, Hustin J. Maternal circulation in the first-trimester human placenta-myth or reality? Am J Obstet Gynecol 1997; 176: 695-705.
- 4. Pijnenborg R, Bland JM, Robertson WB, Brosens I. Uteroplacental arterial changes related to interstitial trophoblast migration in early human pregnancy. Placenta 1983; 4: 397-413.
- 5. Kurjak A, Kupesic S, Hafner T, Kos M, Kostovic-Knezevic L, Grbesa D. Conflicting data on intervillous circulation in early pregnancy. J Perinat Med 1997; 25: 225-36.
- 6. Challier JC. Molécules d’adhérence dans l’implantation et la placentation. In: Merviel P, Challier JC, Foidart JM, Uzan S, eds. Implantation et placentation. Paris: Masson, 2001 : 51-60.
- 7. Hamilton WJ, Boyd JD. Development of the human placenta in the first three months of gestation. J Anat 1960; 94: 297-328.
- 8. Kaufmann P, Benirschke K. Early development of the human placenta. In: Pathology of the human placenta. New York: Springer, 1999 : 42-9.
- 9. Burton GJ, Jauniaux E, Watson AL. Maternal arterial connections to the placental intervillous space during the first trimester of human pregnancy: the Boyd collection revisited. Am J Obstet Gynecol 1999; 181: 718-24.
- 10. Alouini S, Carbillon L, Perrot N, Uzan M, Uzan S. Intervillous and spiral artery flow in normal pregnancies between 5 and 10 weeks of amenorrhea using color doppler ultrasonography. Fetal Diagn Ther 2002; 17: 163-6.
- 11. Carbillon L, Challier JC, Alouini S, Uzan M, Uzan S. Uteroplacental circulation development: Doppler assessment and clinical importance. Placenta 2001; 22: 795-9.
- 12. Damsky CH, Fitzgerald ML, Fisher SJ, Distribution patterns of extracellular matrix components and adhesion receptors are intricately modulated during first trimester cytotrophoblast differentiation along the invasive pathway, in vivo. J Clin Invest 1992; 89: 210-22.
- 13. Goffin F, Maquoi E, Foidart JM. Placentation et métalloprotéases. In: Merviel P, Challier JC, Foidart JM, Uzan S, eds. Implantation et placentation. Paris: Masson, 2001 : 63-94.
- 14. Rodesch F, Simon P, Donner C, Jauniaux E. Oxygen measurements in endometrial and trophoblastic tissues during early pregnancy. Obstet Gynecol 1992; 80: 283-5.
- 15. Jauniaux E, Watson AL, Hempstock J, Bao YP, Skepper JN, Burton GJ. Onset of maternal arterial blood flow and placental oxidative stress. A possible factor in human early pregnancy failure. Am J Pathol 2000; 157: 2111-22.
- 16. Genbacev O, Schubach SA, Miller RK. Villous culture of first trimester human placenta-model to study extravillous trophoblast (EVT) differentiation. Placenta 1992; 13: 439-61.
- 17. Genbacev O, Zhou Y, Ludlow JW, Fisher SJ. Regulation of human placental development by oxygen tension. Science 1997; 277: 1669-72.
- 18. Caniggia I, Mostachfi H, Winter J, et al. Hypoxia-inducible factor-1 mediates the biological effects of oxygen on human trophoblast differentiation through TGFβ3. J Clin Invest 2000; 105: 577-87.
- 19. Caniggia I, Taylor CV, Ritchie JW, Lye SJ, Letarte M. Endoglin regulates trophoblast differentiation along the invasive pathway in human placental villous explants. Endocrinology 1997; 138: 4977-88.
- 20. Huppertz B, Frank HG, Reister F, Kingdom J, Korr H, Kaufmann P. Apoptosis cascade progresses during turnover of human trophoblast: analysis of villous cytotrophoblast and syncytial fragments in vitro. Lab Invest 1999; 79: 1687-702.
- 21. Semenza GL, Agani F, Feldser D, et al. Hypoxia, HIF-1, and the pathophysiology of common human diseases. Adv Exp Med Biol 2000; 475: 123-30.
- 22. Brahimi-Horn C, Berra E, Pouyssegur J. Hypoxia: the tumor’s gateway to progression along the angiogenic pathway. Trends Cell Biol 2001; 11: S32-6.
- 23. Taylor CM, Stevens H, Anthony FW, Wheeler T. Influence of hypoxia on vascular endothelial growth factor and chorionic gonadotrophin production in the trophoblast-derived cell lines: JEG, JAr and BeWo. Placenta 1997; 18: 451-8.
- 24. Grosfeld A, Turban S, Andre J, et al. Transcriptional effect of hypoxia on placental leptin. FEBS Lett 2001; 502: 122-6.
- 25. Jauniaux E, Zaidi J, Jurkovic D, Campbell S, Hustin J. Comparison of colour Doppler features and pathological findings in complicated early pregnancy. Hum Reprod 1994; 9: 2432-7.
- 26. Damsky CH, Fisher SJ. Trophoblast pseudo-vasculogenesis: faking it with endothelial adhesion receptors. Curr Opin Cell Biol 1998; 10: 660-6.
- 27. Chappell L, Bewley S. Pre-eclamptic toxaemia: the role of uterine artery Doppler. Br J Obstet Gynaecol 1998; 105: 379-82.
- 28. Redman CW, Sacks GP, Sargent IL. Preeclampsia: an excessive maternal inflammatory response to pregnancy. Am J Obstet Gynecol 1999; 180: 499-506.
- 29. Zhou Y, Damsky CH, Chiu K, Roberts JM, Fisher SJ. Preeclampsia is associated with abnormal expression of adhesion molecules by invasive cytotrophoblasts. J Clin Invest 1993; 91: 950-60.
- 30. Frendo JL, Therond P, Bird T, et al. Overexpression of copper zinc superoxide dismutase impairs human trophoblast cell fusion and differentiation. Endocrinology 2001; 142: 3638-48.
- 31. Morris NH, Sooranna SR, Learmont JG, et al. Nitric oxide synthase activities in placental tissue from normotensive, pre-eclamptic and growth retarded pregnancies. Br J Obstet Gynaecol 1995; 102: 711-4.
- 32. Granger JP, Alexander BT, Llinas MT, Bennett WA, Khalil RA. Pathophysiology of preeclampsia: linking placental ischemia/hypoxia with microvascular dysfunction. Microcirculation 2002; 9: 147-60.
- 33. Knight M, Redman CW, Linton EA, Sargent IL. Shedding of syncytiotrophoblast microvilli into the maternal circulation in pre-eclamptic pregnancies. Br J Obstet Gynaecol 1998; 105: 632-40.
- 34. Kaufmann P, Luckhardt M, Schweikhart G, Cantle SJ. Cross-sectional features and three-dimensional structure of human placental villi. Placenta 1987; 8: 235-47.
- 35. Krebs C, Macara LM, Leiser R, Bowman AW, Greer IA, Kingdom JC. Intrauterine growth restriction with absent end-diastolic flow velocity in the umbilical artery is associated with maldevelopment of the placental terminal villous tree. Am J Obstet Gynecol 1996; 175: 1534-42.
- 36. Macara L, Kingdom JC, Kaufmann P, et al. Structural analysis of placental terminal villi from growth-restricted pregnancies with abnormal umbilical artery Doppler waveforms. Placenta 1996; 17 : 37-48.
- 37. Kingdom JC, Kaufmann P. Oxygen and placental villous development: origins of fetal hypoxia. Placenta 1997; 18: 613-21.
- 38. Ahmed A, Dunk C, Ahmad S, Khaliq A. Regulation of placental vascular endothelial growth factor (VEGF) and placenta growth factor (PlGF) and soluble Flt-1 by oxygen: a review. Placenta 2000; 21: S16-24.
- 39. Challier JC, Vervelle C, Uzan S. Ontogenesis of villi and fetal vessels in the human placenta. Fetal Diagn Ther 2001; 16: 218-26.
- 40. Challier JC. Environnement et consommation en O2 dans l’unité utéro-foeto-placentaire. Reprod Hum Horm 2002; 4: 225-9.
Liste des figures
Figure 1
Formation des villosités primordiales.
Lors de l’implantation, le trophectoderme cellulaire bordant le blastocyste forme au contact de l’épithélium utérin un syncytiotrophoblaste qui permet l’infiltration du conceptus entier dans l’endomètre à 6 jours post-conception (JPC). Dans ce syncytiotrophoblaste, des lacunes se constituent à 8 JPC et, en fusionnant, créent un espace intervilleux entre les trabécules syncytiales. Le cytotrophoblaste dérivé du trophectoderme prolifère et s’engouffre dans le syncytiotrophoblaste des villosités primordiales (12 JPC) pour former des colonnes cytotrophoblastiques (CC). Ces colonnes de cytotrophoblaste sont envahies par des cellules du mésoderme extra-embryonnaire (14 JPC) où apparaissent les premiers vaisseaux foetaux vers 18 JPC. Le mésoderme repousse les cellules cytotrophoblastiques à la base des villosités donnant naissance au cytotrophoblaste extravillositaire (CEV). Certaines villosités qui atteignent l’endomètre sont appelées villosités crampons (VC), les autres sont dites flottantes (VF) et elles présentent parfois des îlots cytotrophoblastiques (IC).
Figure 2
Circulation intervilleuse au premier trimestre.
À 8 semaines d’aménorrhée, les artères spiralées de la décidue sont partiellement bouchées par le cytotrophoblaste extravillositaire (CEV). Le plasma et quelques hématies maternelles circulent dans l’espace intervilleux (flèches jaunes). Le sang est court-circuité vers un réseau de sinusoïdes déciduaux et regagne les veines. À 12 semaines d’aménorrhée, les bouchons trophoblastiques ont disparu et le sang des artères utéro-placentaires s’écoule dans l’espace intervilleux. Le rôle des sinusoïdes déciduaux est devenu insignifiant. pO2: pression partielle d’oxygène.
Figure 3
Invasion trophoblastique (8e semaine d’aménorrhée).
La paroi musculo-élastique des artères spiralées est remodelée du fait de l’invasion par le cytotrophoblaste extravillositaire, en provenance de la base des villosités crampons. Celui-ci dissocie la paroi musculo-élastique des artères et se substitue par endroits à l’endothélium. Au cours de l’invasion de la décidue et de ses vaisseaux, le cytotrophoblaste acquiert des phénotypes variés qui se traduisent par l’expression de marqueurs cytologiques spécifiques. Différentes sous-unités d’intégrines sont observées selon que le cytotrophoblaste est de type épithélial (α3β4, α6β4), interstitiel (α1β3, α5β1) ou endovasculaire (α1β3, αVβ3). Le cytotrophoblaste met également en oeuvre des métalloprotéases matricielles (MMP) synthétisées et activées au cours de la progression des cellules dans la matrice extracellulaire comme la gélatinase B (MMP9) qui remplace la gélatinase A (MMP2). MIB-1: anticorps détectant KI67 en phase G1, S, G2 et M du cycle cellulaire ; TGFβ3: transforming growth factor β3; cadhérines E et V-E: molécules d’adhérence; HIF1α: hypoxia inducible factor-1α; MMP1, 2, 9: matrix metalloproteinase 1, 2, 9; PECAM-1: platelet-endothelial cell adhesion molecule-1.
Figure 4
Pression partielle d’oxygène et expression des gènes dans le trophoblaste.
Plusieurs facteurs de transcription inductibles par l’hypoxie, HIF 1, 2 et 3 ont été mis en évidence. Dans le cas d’HIF1, la sous-unité HIF1α est stable en hypoxie et, après translocation dans le noyau, forme un hétérodimère avec la sous-unité HIF1. La liaison du dimère HIF1 à une séquence régulatrice HRE (consensus 5’-RCGTG-3’) de l’ADN avec d’autres co-facteurs induit la transcription de gènes contrôlés par l’O2: VEGF (vascular endothelial growth factor), Glut1 (glucose transporter 1), leptine, etc. L’activation des gènes à séquence HRE induit directement ou indirectement la prolifération trophoblastique. Certains membres de la famille du TGFβ comme TGFβ2, présentent une séquence d’ADN HRE fixant HIF1α. TGFβ3 pourrait présenter cette séquence et son effet s’exercerait en inhibant la différenciation. En normoxie, la prolyl-hydroxylase activée hydroxyle la proline 564 du domaine carboxy-terminal de l’HIF1α. Celui-ci fixe alors le facteur suppresseur de tumeur de von Hippel-Lindau, qui associé à d’autres éléments, forme un complexe ayant une activité E3 ubiquitine ligase. L’HIF1α peut alors se lier à des ubiquitines et il est dégradé par le protéasome. D’autres capteurs d’O2 à groupement hémique agissant sur HIFα ont été proposés.
Figure 5
Physiopathogénie possible de la pré-éclampsie.
La cause initiale de la pré-éclampsie n’est pas clairement identifiée mais la séquence des événements qui la déclenchent commence à être connue. Du fait d’une invasion trophoblastique incomplète, les artères spiralées conserveraient leur assise musculo-élastique dans leur segment myométrial et parfois endométrial, entraînant une perfusion inadéquate de l’espace intervilleux au premier trimestre. Le remodelage défectueux des artères spiralées provoque ensuite des altérations récurrentes de la circulation utéro-placentaire dues à la production d’ion superoxyde (O.2) au cours du stress oxydatif. Le dysfonctionnement de l’endothélium déclenche aussi une production accrue d’agents vasoconstricteurs (thromboxane et endothéline). L’expression de certaines cytokines pro-inflammatoires et immunorégulatrices produites au cours du stress oxydatif, comme le TNFα (tumor necrosis factor α) et les interleukines IL-1α et IL-10, est augmentée dans les placentas pré-éclamptiques et les concentrations de TNFα et d’IL-6 s’élèvent dans le sang maternel. On observe aussi dans le sang maternel un dérèglement du système de coagulation sanguine favorisant l’agrégation plaquettaire. Le mécanisme de diffusion des altérations placentaires à la mère reste obscur. L’excès de microfragments nécrotiques ou apoptotiques de syncytiotrophoblaste dans la circulation maternelle pourrait conduire à un dysfonctionnement généralisé de l’endothélium des vaisseaux maternels et participer ainsi à l’amplification de la réaction inflammatoire prédisposant à l’hypertension. RCIU: retard de croissance intra-utérin.
Figure 6
Environnement en oxygène et maladies.
L’oxygène peut aussi affecter l’ontogenèse des villosités foetales dont les phénotypes diffèrent selon les maladies et leur degré de gravité. Un aspect hypoxique (hyper-ramification et hyper-capillarisation des villosités terminales) a été noté dans la pré-éclampsie. Un aspect hyperoxique (ramification villositaire déficiente associée à une angiogenèse) prédomine dans la pré-éclampsie avec retard de croissance intra-utérin (RCIU) associée à une vélocité diastolique absente ou inversée (VDAI) ou dans le RCIU sans pré-éclampsie avec VDAI. L’anémie maternelle et la grossesse en altitude sont à l’origine de deux situations où l’appauvrissement en O2 a des répercussions caractéristiques sur la morphogenèse et l’angiogenèse villositaires. Dans les grossesses en altitude, on observe une ramification très importante des villosités terminales et une hyper-capillarisation.