Corps de l’article
La progression tumorale est classiquement décrite comme un processus que caractérise l’expansion de clones cellulaires différents d’où émerge un clone de cellules métastatiques au caractère agressif, conséquence de la sélection positive de cellules ayant accumulé des altérations génétiques dans la tumeur. Étape tardive de la progression tumorale, la dissémination de cellules tumorales à partir du foyer initial va conduire, in fine, à la formation de métastases spécifiques à certains tissus selon le concept du seed and soil, au prix de processus de sélection et d’expansion de quelques cellules ayant acquis des propriétés supplémentaires et essentielles à la colonisation tissulaire [1].
Deux modèles se sont affrontés très récemment encore pour expliquer l’origine du potentiel métastatique d’une tumeur primitive: celui-ci peut résulter soit de la sélection progressive de très rares cellules variantes dotées de ce potentiel, soit au contraire, l’ensemble des cellules tumorales serait d’emblée doté de cette prédisposition. Au niveau génique, le premier modèle attribue un rôle majeur aux «gènes suppresseurs de métastases» dont la déficience provoque l’expression du caractère métastatique; le second stipule que les oncogènes et/ou les gènes suppresseurs de tumeur – dont les altérations déterminent à la fois l’état transformé et la progression tumorale – sont les acteurs à part entière de la dissémination métastatique [2]. Faiblement étayé par des arguments expérimentaux, ce vieux débat s’est ravivé l’an dernier [2, 3] à la faveur des premiers résultats du typage moléculaire des cancers.
Rappelons que les profils d’expression de tumeurs solides obtenus à l’aide de la technologie des microarrays avaient rapidement permis de distinguer des cancers de bon et de mauvais pronostic. En particulier, les travaux de Friend et R. Bernards [4] ont identifié un groupe de 70 gènes dont l’expression caractérise un groupe de cancers du sein «prédisposés» au processus métastatique, alors qu’aucune métastase n’était détectable chez les patientes lors de l’exérèse chirurgicale de leur tumeur. Mieux encore ! Une «signature génique d’un potentiel métastatique» commune à plusieurs types de tumeurs cancéreuses a été identifiée [5].
Cette «signature transcriptionnelle» du caractère métastatique d’une tumeur a été obtenue en deux temps, en croisant d’ailleurs les résultats obtenus par deux plate-formes de génomique utilisant les technologies Affymetrix et Rosetta. Tout d’abord la comparaison de l’expression génique de populations cellulaires provenant de tumeurs primitives épithéliales prélevées au moment du diagnostic et de leurs métastases a révélé une expression différentielle de 128 gènes (hypo- ou hyper-exprimés dans les métastases par rapport à la tumeur primaire), une signature complexe mais heuristique puisque retrouvée dans certaines tumeurs primaires; cette signature moléculaire caractéristique d’un pouvoir métastatique a été vérifiée en analysant les cellules de 279 tumeurs solides primitives d’origine diverse. De cette analyse a émergé un profil génique intéressant 17 gènes, et caractéristique des cancers ayant métastasé et/ou à potentialité métastatique. Dans le cas des cancers de la prostate et du médulloblastome, cette « signature » moléculaire était associée à un mauvais pronostic, et dans le cas des cancers du sein, elle indiquait une propension à métastaser. Ainsi l’évolution d’un cancer pourrait-elle être déterminée précocement, si la population tumorale primitive est constituée essentiellement de cellules ayant d’emblée un potentiel métastatique, et, dans ce cas, elle ne s’expliquerait pas par la sélection stochastique de quelques rares cellules. En revanche, dans le cas des lymphomes diffus à grandes cellules B, cette signature n’était pas prédictive du pronostic. Avant de développer un test pronostique clinique fondé sur ces données moléculaires, attendons l’analyse d’un plus grand nombre de tumeurs. Remarquons qu’aucun des 17 gènes sélectionnés (huit gènes hyper-exprimés, neuf hypo-exprimés, Tableau I) ne code pour des molécules directement impliquées dans le processus métastatique. Parmi les gènes hyper-exprimés, quatre codent pour des composants de la machinerie de traduction protéique, deux sont essentiels pour l’expression du collagène I, enfin l’un d’entre eux code pour la Sécurine, une protéine impliquée dans la régulation de la séparation des chromatides soeurs et qui module négativement l’activité de p53 [6] ((→) m/s 2001, n° 3, p.353). Rappelons que ce groupe de gènes a été identifié à partir d’un tissu tumoral entier très hétérogène. Beaucoup des protéines codées par ces gènes sont impliquées dans le dialogue stroma-tumeur caractérisé par les éléments stromaux activés du tissu tumoral.
Tableau I
Les 17 gènes associés au caractère métastatique.
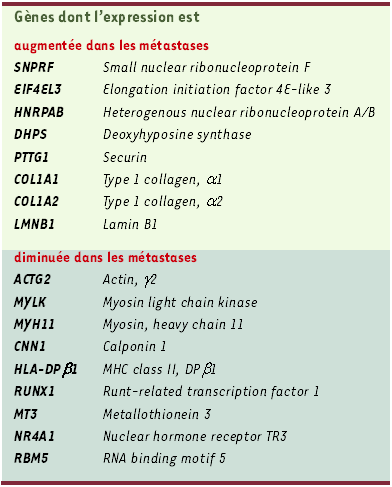
(adapté de [5])
Quoi qu’il en soit, ce travail est vraiment pionnier en ce qu’il propose qu’un tout petit nombre de gènes révèle d’emblée le potentiel métastatique d’une tumeur primitive.
Tout récemment, le groupe de J. Massagué a réalisé l’analyse génomique fonctionnelle de cellules d’une lignée représentative d’un modèle de cancer mammaire humain (lignée MDA-MB-231) [7], capable de dissémination osseuse chez la souris. La lignée parentale (dérivée de tissus métastatiques prélevés chez une patiente) contient bien la signature de 70 gènes associée à un mauvais pronostic (et donc au potentiel métastatique) décrite antérieurement selon le raisonnement récurrent des auteurs [4]. Mais à cet ensemble caractéristique s’ajoute, dans les lignées à haut potentiel métastatique osseux du modèle, l’expres- sion de 5 gènes propres à l’invasion du tissu osseux (CXCR-4, IL-11, CTGF, MMP1[voir brève de ce numéro, p. 1078], Ostéopontine)[1] dont la co-expression est associée à une forte capacité ostéolytique.
À la lumière de toutes ces données constatons que, par sa puissance à analyser la complexité, la génomique fonctionnelle permet de résoudre l’apparent conflit des deux modèles: plus que de s’opposer, ils se complètent et se chevauchent. En effet, si toutes les cellules malignes de la tumeur primitive ont une capacité de dissémination à distance, seules des cellules variantes, rares, établiront des métastases dans des sites tissulaires spécifiques pour chaque cancer.
Parties annexes
Note
-
[1]
CXCR4: ligand de la chimiokine SDF-1/CXCL12, stromal derived factor; CTGF: connective tissue-derived growth factor; MMP1: matrix metalloproteinase 1.
Références
- 1. Couzin J. Tracing the steps of metastasis, cancer’s menacing ballet. Science 2003; 299: 1002-6.
- 2. Bernards R, Weinberg R. A progression puzzle. Nature 2002; 418: 523.
- 3. Edwards P. Metastasis: the role of chance in malignancy. Nature 2002; 419: 559.
- 4. Van de Vijver MJ, He YD, Van’t Veer LJ, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med 2002; 347: 1999-2009.
- 5. Ramaswamy S, Ross K, Lander E, Golub T. A molecular signature of metastasis in primary solid tumors. Nat Genet 2003; 33: 49-54
- 6. Bernal JA, Luna R, Espina A, et al. Human securin interacts with p53 and modulates p53-mediated transcriptional activity and apoptosis. Nat Genet 2002; 32: 306-11.
- 7. Kang Y, Siegel PM, Shu W, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003; 3: 537-49.
Liste des tableaux
Tableau I
Les 17 gènes associés au caractère métastatique.
(adapté de [5])