Corps de l’article
L’élucidation des bases génétiques de la prédisposition au développement de certains cancers constitue depuis ces dernières années un enjeu majeur pour tenter d’aborder des mécanismes fondamentaux impliqués dans les processus de cancérogenèse. Le syndrome de Bloom, maladie génétique rare, autosomique et récessive, présente l’une des meilleures corrélations connues entre instabilité génétique et prédisposition tumorale, et notre équipe se consacre à l’étude de cette maladie. Du fait du mode de transmission récessif de cette maladie, nous nous posions la question de savoir si le gène BLM, dont les mutations sont à l’origine de ce syndrome, pourrait jouer un rôle dans le développement de cancers dans la population générale. La réponse est oui, comme en témoignent deux études publiées très récemment dans Science [1, 2]. En effet, ces travaux montrent que le fait d’être porteur d’une mutation sur une seule des deux copies du gène BLM est suffisant pour accroître le risque de développer une tumeur colorectale chez l’homme et chez la souris.
La mutation des deux copies du gène BLM est à l’origine du syndrome de Bloom (BS) dont la caractéristique clinique majeure est l’incidence élevée de tumeurs. En effet, ces patients présentent une prédisposition à développer tous les types de cancers affectant la population générale mais à un âge plus précoce (Tableau I) [3]. De plus, les cellules dérivées de patients BS sont caractérisées par un phénotype mutateur et une instabilité génétique généralisée se manifestant notamment par une augmentation de 10 fois du taux d’échanges entre chromatides soeurs qui constitue le seul critère diagnostic objectif de la maladie [4]. Le gène BLM est localisé sur le chromosome 15 en position 15q26.1 et code pour la protéine BLM de 1417 acides aminés. Cette protéine appartient à la sous-famille des hélicases RecQ [5] ((→) m/s 2002, n°1, p. 79) qui sont caractérisées par la présence, dans leur région centrale, d’un domaine contenant sept motifs hélicase consensus très bien conservés. À ce jour, neuf mutations différentes du gène BLM ont été décrites dont quatre conduisent à la formation de protéines tronquées ne possédant plus de domaine hélicase consensus. Une de ces mutations, la mutation BLMAsh, crée un codon stop prématuré au niveau de l’exon 10 et a été retrouvée à l’état homozygote chez quatre patients non apparentés d’origine juive ashkénaze, ce qui a confirmé un effet fondateur dans cette population [5].
Tableau I
Tableau récapitulatif des 100 tumeurs affectant 71 patients BS parmi les 168 répertoriés dans le registre du syndrome de Bloom au 1er juillet 1996.
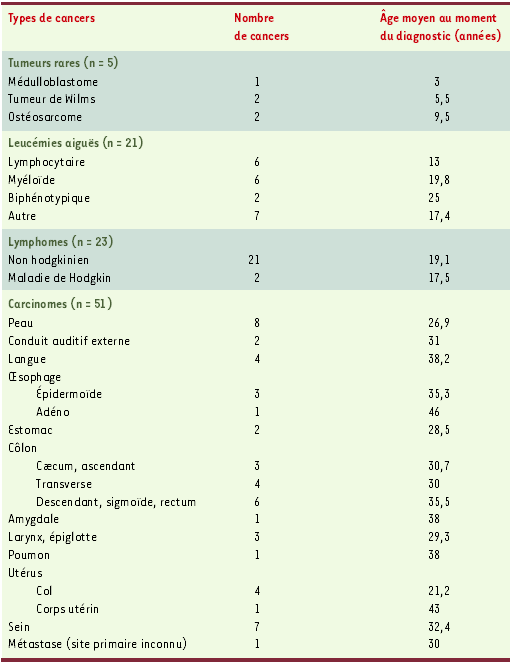
(d’après [3])
L’étude publiée par Gruber et al. [1] montre que des individus porteurs de la mutation BLMAsh à l’état hétérozygote (environ 1 % des individus d’origine juive ashkénaze) ont un risque plus de deux fois plus élevé que la population témoin de développer un cancer colorectal. Ces résultats ont été reproduits par l’équipe de Joanna Groden dans un modèle murin [2]. En effet, cette équipe a développé des souris chez lesquelles une copie de l’homologue murin du gène BLM a été invalidée, produisant ainsi un allèle mutant BlmCin. Des souris BlmCin/+ hétérozygotes pour cet allèle ont été croisées avec des souris ApcMin/+ hétérozygotes pour une mutation dans le gène suppresseur de tumeur Apc, les prédisposant au développement de multiples adénomes intestinaux et représentant un modèle de la polypose adénomateuse familiale humaine. Les souris ApcMin/+ BlmCin/+ issues de ce croisement développent deux fois plus de tumeurs intestinales que les souris témoins ApcMin/+ Blm+/+, et ces tumeurs présentent une perte de l’allèle Apc sauvage, sans perte de l’allèle Blm sauvage. Cette étude [2] suggère que l’haplo-insuffisance du gène Blm pourrait suffire à favoriser la formation de tumeurs. Cependant, bien que la protéine Blm soit détectable dans les tumeurs intestinales des souris ApcMin/+ BlmCin/+, on ne peut formellement exclure que l’allèle Blm sauvage n’ait été inactivé par une mutation ponctuelle n’altérant ni la taille ni le taux d’expression de la protéine, mais conférant le phénotype mutateur caractéristique des cellules BS. C’est pourquoi l’analyse du taux d’échanges entre chromatides soeurs dans les cellules de tumeurs colorectales dérivées de patients hétérozygotes pour la mutation BLMAsh (et/ou de souris ApcMin/+ BlmCin/+) donnerait des informations précieuses quant au mécanisme de développement de ces tumeurs. Il serait également intéressant de savoir si les souris BlmCin/+ développent spontanément plus de tumeurs que les souris sauvages, comme cela a récemment été montré chez des souris hétérozygotes pour une mutation du gène ATM, impliqué dans un autre syndrome autosomique récessif associant instabilité génétique et cancer, l’ataxie-télangiectasie [6].
Mais si ces tumeurs sont effectivement la conséquence d’une haplo-insuffisance du gène BLM, la question se pose alors de savoir pourquoi un allèle mutant du gène BLM est récessif pour le développement de la maladie et d’une instabilité génétique majeure, et partiellement dominant pour le développement de cancers. En effet, les individus hétérozygotes pour une mutation du gène BLM ne manifestent aucune des caractéristiques cliniques associées au syndrome de Bloom, et leurs cellules ne présentent pas d’élévation du taux d’échanges entre chromatides soeurs. L’explication la plus probable serait que la quantité de protéine BLM produite par une seule copie du gène BLM soit suffisante pour la majorité de ses fonctions. Cependant, dans certaines conditions, peut-être en réponse à des lésions de l’ADN, la quantité de protéine BLM pourrait devenir limitante. En effet, bien que la fonction de la protéine BLM soit encore mal définie, plusieurs études suggèrent un rôle essentiel de cette 3’-5’ ADN hélicase [7] dans des mécanismes de surveillance du génome [8], de résolution de structures anormales de l’ADN [9, 10], et/ou de réparation de lésions de l’ADN, notamment en réponse à des stress génotoxiques [11, 12]. Si la protéine BLM est impliquée dans de tels processus au sein de complexes protéiques, et dans le cadre d’interactions stoechiométriques bien déterminées, alors une réduction de la quantité de protéine BLM pourrait être délétère pour la cellule. Mais l’inactivation d’une copie du gène BLM pourrait ne pas systématiquement résulter en une haplo-insuffisance, au moins chez la souris, car les résultats présentés par l’équipe de J. Groden [2] diffèrent de ceux publiés il y a deux ans par une autre équipe [13]. En effet, Luo et al. avaient développé un modèle murin du syndrome de Bloom en invalidant les 2 copies du gène Blm. Des souris hétérozygotes Blm-/+avaient été croisées, comme dans l’étude de Joanna Groden, avec des souris ApcMin/+, mais les souris ApcMin/+ Blm-/+issues de ce croisement ne présentaient pas d’augmentation du nombre de tumeurs intestinales par rapport aux souris témoins. Cette contradiction pose fondamentalement la question du mécanisme et/ou du contexte génétique conférant une prédisposition au développement de cancers à la suite de l’inactivation d’une copie du gène BLM.
Il n’en demeure pas moins que ces nouvelles données [1, 2] ouvrent d’importantes perspectives aussi bien pour la mise en place d’un conseil génétique pour des patients atteints d’un cancer et ayant un ancêtre d’origine juive ashkénaze que, sur un plan plus fondamental, pour l’étude du rôle que pourrait jouer le gène BLM dans des processus de cancérogenèse dans la population générale.

Parties annexes
Références
- 1. Gruber SB, Ellis NA, Rennert G, et al. BLM heterozygosity and the risk of colorectal cancer. Science 2002; 297: 2013.
- 2. Goss KH, Risinger MA, Kordich JJ, et al. Enhanced tumor formation in mice heterozygous for Blm mutation. Science 2002; 297: 2051-3.
- 3. German J. Bloom’s syndrome XX. The first 100 cancers. Cancer Genet Cytogenet 1997; 93: 100-6.
- 4. German J. Bloom syndrome: a mendelian prototype of somatic mutational disease. Medicine(Baltimore) 1993; 72: 393-406.
- 5. Ellis NA, Groden J, Ye TZ, et al. The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell 1995; 83: 655-66.
- 6. Spring K, Ahangari F, Scott SP, et al. Mice heterozygous for mutation in Atm, the gene involved in ataxia-telangiectasia, have heightened susceptibility to cancer. Nat Genet 2002; 32: 185-90.
- 7. Karow JK, Chakraverty RK, Hickson ID. The Bloom’s syndrome gene product is a 3’-5’ DNA helicase. J Biol Chem 1997; 272: 30611-4.
- 8. Wang Y, Cortez D, Yazdi P, Neff N, Elledge SJ, Qin J. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev 2000; 14: 927-39.
- 9. Karow JK, Constantinou A, Li JL, West SC, Hickson ID. The Bloom’s syndrome gene product promotes branch migration of holliday junctions. Proc Natl Acad Sci USA 2000; 97: 6504-8.
- 10. Huber MD, Lee DC, Maizels N. G4 DNA unwinding by BLM and Sgs1p: substrate specificity and substrate-specific inhibition. Nucleic Acids Res 2002; 30: 3954-61.
- 11. Ababou M, Dutertre S, Lecluse Y, Onclercq R, Chatton B, Amor-Gueret M. ATM-dependent phosphorylation and accumulation of endogenous BLM protein in response to ionizing radiation. Oncogene 2000; 19: 5955-63.
- 12. Dutertre S, Sekhri R, Tintignac LA, et al. Dephosphorylation and subcellular compartment change of the mitotic Bloom’s syndrome DNA helicase in response to ionizing radiation. J Biol Chem 2002; 277: 6280-6.
- 13. Luo G, Santoro IM, McDaniel LD, et al. Cancer predisposition caused by elevated mitotic recombination in Bloom mice. Nat Genet 2000; 25: 424-9.
Liste des tableaux
Tableau I
Tableau récapitulatif des 100 tumeurs affectant 71 patients BS parmi les 168 répertoriés dans le registre du syndrome de Bloom au 1er juillet 1996.
(d’après [3])