Corps de l’article
Les syndromes myéloprolifératifs chroniques (SMP) sont des hémopathies malignes ayant pour caractéristiques communes une hyperplasie myéloïde globale et une hypersensibilité aux cytokines des progéniteurs hématopoïétiques. Ce sont des maladies acquises, clonales, de la cellule souche hématopoïétique. La classification des SMP distingue : la leucémie myéloïde chronique (LMC), la polyglobulie primitive ou maladie de Vaquez, la thrombocytémie essentielle, la splénomégalie myéloïde ou myélofibrose primitive, le syndrome hyperéosinophilique, et des SMP atypiques [1].
La physiopathologie de la LMC est liée à l'existence d'une translocation équilibrée spécifique, la translocation t(9 ; 22), à l'origine du chromosome Philadelphie, qui engendre le gène de fusion BCR-ABL. La protéine bcr-abl possède une activité tyrosine kinase constitutive et interagit avec de nombreuses voies de signalisation intracellulaires. Ces dernières années, le développement d'un médicament capable d'inhiber l'activité tyrosine kinase de bcr-abl, l'imatinib mésylate (Glivec®), a profondément modifié la prise en charge thérapeutique de cette leucémie [2]. Dans les SMP atypiques, plusieurs anomalies moléculaires ont été identifiées, conduisant toujours à une activation constitutive d'une protéine à activité tyrosine kinase [3]. Récemment, une protéine de fusion à activité tyrosine kinase constitutive a été mise en évidence dans les syndromes hyperéosinophiliques, liée à une délétion interstitielle située sur le chromosome 4 en 4q12. L'imatinib est remarquablement efficace dans les syndromes hyperéosinophiliques [4], ainsi que dans certains SMP atypiques [5, 6].
En l'absence de translocation chromosomique récurrente dans la polyglobulie de Vaquez, l'anomalie moléculaire à l'origine de la maladie est restée inconnue jusqu'ici [7, 8], mais, par analogie avec la LMC, il était probable que l'activation anormale d'une tyrosine kinase puisse être impliquée. Les progéniteurs de polyglobulie de Vaquez présentent une hypersensibilité à l'érythropoïétine (Epo), ainsi qu'au SCF (stem-cell factor), au GM-CSF (granulocyte-macrophage-colony stimulating factor), à l'IL-3 (interleukine-3) et à l'IGF-1 (insulin-like growth factor-1). En utilisant des inhibiteurs biochimiques, nous avons montré que les voies JAK2-STAT5 et PI3-kinase étaient nécessaires à la différenciation érythroblastique terminale indépendante de l'Epo [9].
JAK2 est une protéine à activité tyrosine kinase constitutivement associée au récepteur de l'Epo, dépourvu lui-même d'activité kinase intrinsèque. Dans des conditions physiologiques, la phosphorylation de JAK2 induite après fixation de l'Epo sur son récepteur est la première étape nécessaire à l'activation des voies de signalisation. Nous avons mis en évidence, par séquençage direct de la partie codante du gène JAK2, une mutation ponctuelle située dans l'exon 12, présente chez environ 85 % des polyglobulies de Vaquez étudiées. La mutation n'a été retrouvée chez aucun des témoins étudiés (normaux et polyglobuliques secondaires). Cette mutation entraîne la substitution d'une valine en phénylalanine en position 617 (V617F). Il s'agit d'une mutation unique (aucune autre mutation de JAK2 n'a été trouvée), et clonale car elle est présente dans les cellules myéloïdes des patients (PNN, érythroblastes, plaquettes), mais pas dans leurs lymphocytes T [10].
La mutation JAK2 V617F est située dans le domaine JH2 ou domaine pseudo-kinase de JAK2, qui régule négativement l'activité kinase de la protéine. Sur un plan fonctionnel, la protéine JAK2 mutée est spontanément active, capable d'induire une activité transcriptionnelle de STAT5 en l'absence d'Epo dans un modèle de transfection transitoire, et de conférer une hypersensibilité aux cytokines dans des lignées cellulaires (Figure 1A). Les effets in vivo de la mutation ont été étudiés. Des souris greffées avec des cellules de moelle osseuse exprimant la protéine JAK2 mutée développent une polyglobulie et une splénomégalie (Figure 1B). L'ensemble de ces résultats nous permet de proposer que la mutation JAK2 V617F est l'événement moléculaire primaire et suffisant pour entraîner le développement d'une polyglobulie primitive [10].
Figure 1
La protéine JAK2 mutée entraîne une hypersensibilité à l'érythropoïétine in vitro et une polyglobulie in vivo.
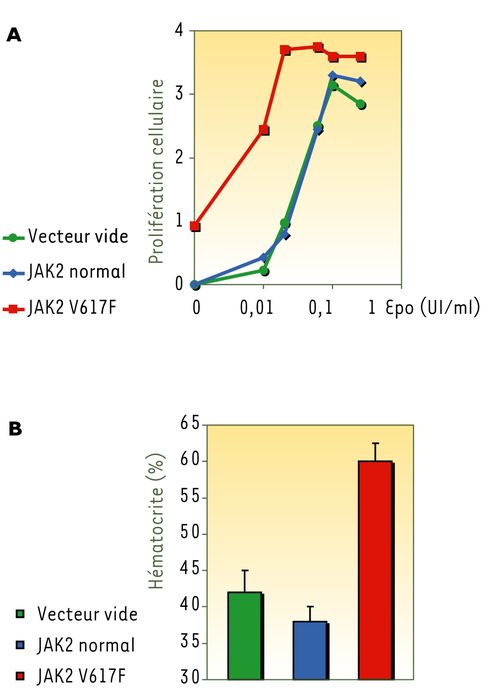
A. Des lignées cellulaires dépendantes des cytokines ont été infectées par un rétrovirus codant pour JAK2 V617F : une partie de ces cellules devient indépendante des cytokines (Epo) alors que les cellules témoins meurent. De plus, les cellules exprimant JAK2 V617F deviennent hypersensibles aux cytokines, réalisant ainsi un « phénotype cellulaire » très proche de celui observé chez les patients. B. Des souris irradiées ont été greffées avec de la moelle osseuse infectée par le rétrovirus codant pour JAK2 V617F : elles développent une polyglobulie et une splénomégalie quatre semaines après la greffe.
De façon surprenante pour une mutation activatrice, la mutation est retrouvée à l'état homozygote chez environ 30 % des polyglobulies de Vaquez. Cela est lié à une perte d'hétérozygotie du bras court du chromosome 9 (où est situé JAK2), secondaire à un phénomène de recombinaison mitotique [11]. Pourquoi une mutation gain de fonction s'accompagne-t-elle d'une perte d'hétérozygotie, phénomène qui n'est habituellement retrouvé que pour les gènes suppresseurs de tumeur ? Nous avons montré que la perte du gène JAK2 normal confère un avantage prolifératif aux cellules [10], mais d'autres questions restent en suspens.
La découverte de cette mutation unique de JAK2 a été rapportée par cinq équipes [10, 12-15]. Tous ces travaux confirment que la mutation est retrouvée dans la grande majorité des cas de polyglobulie primitive (65 % à 97 % selon les études), mais également dans 25 % à 50 % des thrombocytémies essentielles et des splénomégalies myéloïdes.
L'identification de la mutation JAK2 V617F représente une avancée majeure dans la compréhension des syndromes myéloprolifératifs non-LMC, même si les mécanismes par lesquels une mutation unique aboutit à des phénotypes différents restent à élucider [16-18]. Sur un plan médical, la découverte de cette mutation permet pour la première fois de réaliser un test diagnostique moléculaire de ces maladies. Enfin, quelques années après la mise sur le marché du Glivec®, les perspectives de thérapie ciblée, ouvertes en hématologie comme en cancérologie par les inhibiteurs de tyrosine kinase [5, 6], permettent d'espérer à moyen terme la mise au point d'un médicament spécifique des syndromes myéloprolifératifs JAK2 V617F.
Parties annexes
Références
- 1. Vardiman JW, Harris NL, Brunning RD. The World Health Organization (WHO) : classification of myeloid neoplasms. Blood 2002 ; 100 : 2292-302.
- 2. Wong S, Witte ON. The BCR-ABL story : bench to bedside and back. Annu Rev Immunol 2004 ; 22 : 247-306.
- 3. Cross NC, Reiter A. Tyrosine kinase fusion genes in chronic myeloproliferative diseases. Leukemia 2002 ; 16 : 1207-12.
- 4. Gotlib J, Cools J, Malone JM, et al. The FIP1L1-PDGFRalpha fusion tyrosine kinase in hypereosinophilic syndrome and chronic eosinophilic leukemia implications for diagnosis, classification and management. Blood 2004 ; 103 : 2879-91.
- 5. Pardanani A, Tefferi A. Imatinib targets other than bcr/abl and their clinical relevance in myeloid disorders. Blood 2004 ; 104 : 1931-9.
- 6. Wadleigh M, De Angelo DJ, Griffin JD, Stone RM. After chronic myelogenous leukemia : tyrosine kinase inhibitors in others malignancies. Blood 2005 ; 105 : 22-30.
- 7. Pahl HL. Towards a molecular understanding of polycythemia rubra vera. Eur J Biochem 2000; 267: 3395-401.
- 8. Spivak JL. Polycythemia vera : myths, mechanisms, and management. Blood 2002 ; 100 : 4272-90.
- 9. Ugo V, Marzac C, Teyssandier I, et al. Multiple signaling pathways are involved in erythropoietin-independent differentiation of erythroid progenitors in polycythemia vera. Exp Hematol 2004 ; 32 : 179-87.
- 10. James C, Ugo V, Le Couedic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005 ; 434 : 1144-8.
- 11. Kralovics R, Guan Y, Prchal JT. Acquired uniparental disomy of chromosome 9p is a frequent stem cell defect in polycythemia vera. Exp Hematol 2002 ; 30 : 229-36.
- 12. Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005 ; 365 : 1054-61.
- 13. Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 2005 ; 352 : 1779-90.
- 14. Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005 ; 7 : 387-97.
- 15. Zhao R, Xing S, Li Z, et al. Identification of an acquired JAK2 mutation in Polycythemia vera. J Biol Chem 2005 online.
- 16. Goldman JM. A unifying mutation in chronic myeloproliferative disorders. N Engl J Med 2005 ; 352 : 1744-6.
- 17. Kaushansky K. On the molecular origins of the chronic myeloproliferative disorders : it all makes sense. Blood 2005 ; 105 : 4187-90.
- 18. Shannon K, Van Etten RA. JAKing up hematopoietic proliferation. Cancer Cell 2005 ; 7 : 291-3.
Liste des figures
Figure 1
La protéine JAK2 mutée entraîne une hypersensibilité à l'érythropoïétine in vitro et une polyglobulie in vivo.
A. Des lignées cellulaires dépendantes des cytokines ont été infectées par un rétrovirus codant pour JAK2 V617F : une partie de ces cellules devient indépendante des cytokines (Epo) alors que les cellules témoins meurent. De plus, les cellules exprimant JAK2 V617F deviennent hypersensibles aux cytokines, réalisant ainsi un « phénotype cellulaire » très proche de celui observé chez les patients. B. Des souris irradiées ont été greffées avec de la moelle osseuse infectée par le rétrovirus codant pour JAK2 V617F : elles développent une polyglobulie et une splénomégalie quatre semaines après la greffe.