Résumés
Résumé
Dans la nature, certaines espèces, animales comme végétales, se reproduisent par parthénogenèse, c’est-à-dire uniquement à partir du gamète femelle, sans participation du gamète mâle. Chez les mammifères, la parthénogenèse naturelle n’a jamais été observée, ce qui suggère que le développement d’un embryon de mammifère requiert la présence des deux génomes maternel et paternel. Cela est dû à un phénomène, découvert au début des années 1980, appelé « empreinte génomique parentale » : il apparaît que, chez tous les mammifères, les génomes mâle et femelle qui se rencontrent dans l’oeuf fécondé sont marqués d’un sceau différent, nommé empreinte. Par la suite, l’identification de gènes spécifiques soumis à empreinte parentale a permis de montrer que cette empreinte conduit à une expression monoallélique, dépendante de l’origine parentale. Les caractéristiques moléculaires de ce phénomène de marquage épigénétique ont maintenant été décrites et permettent d’expliquer certaines maladies humaines liées à des gènes soumis à empreinte.
Summary
Genomic imprinting leads to parent-of-origin-specific monoallelic expression of about 60 known genes in the mammalian genome. It was discovered 20 years ago and the aim of this review is to summarize its main characteristics. The nature of the imprint, still unknown, is characterized by differential chromatin structure and DNA methylation. The imprint is reset at each generation during gametogenesis, which can be observed by demethylation in the PGCs, then gamete-specific remethylation. The imprinted genes are usually located in clusters and regulated by cis sequences such as imprinting centres, trans factors such as the insulator protein CTCF and/or large non coding antisense RNAs. Genetic and epigenetic abnormalities of the imprinted clusters can lead to human diseases such as Prader-Willi, Angelman or Beckwith-Wiedemann syndromes.
Corps de l’article
Découverte de l’empreinte parentale
En 1984, Davor Solter mit au point une technique de transfert nucléaire permettant d’échanger, dans des oeufs de souris fécondés, les noyaux issus du spermatozoïde et ceux issus de l’ovocyte. Des gynogénotes (deux noyaux femelles) ou des androgénotes (deux noyaux mâles) ont ainsi été produits, mais ces embryons ne se développaient jamais à terme, malgré leur composition diploïde [1, 2]. Cette expérience permit d’évaluer le rôle respectif des deux génomes : les gynogénotes présentaient un développement à peu près normal de l’embryon lui-même, tandis que les annexes embryonnaires étaient anormales ; la situation inverse était observée pour les androgénotes (embryon anormal, annexes normales). Les génomes mâle et femelle semblaient donc jouer un rôle symétrique et complémentaire. Ces observations révélèrent également l’existence d’un phénomène de marquage des génomes parentaux, appelé empreinte génomique parentale (genomic imprinting). Ainsi, les génomes maternel et paternel, malgré leur constitution génique identique, ne fonctionnent pas de façon équivalente, car ils sont marqués par cette empreinte, apposée durant la gamétogenèse. Il en résulte une différence fonctionnelle des deux génomes parentaux, qui rend la présence de chacun d’entre eux indispensable au développement à terme de l’embryon.
Par la suite, des études génétiques furent réalisées à partir de croisements de souris portant des remaniements chromosomiques : ces croisements permettaient d’obtenir des individus chez lesquels tout ou partie d’une paire de chromosomes provenait d’un seul des deux parents (unidisomie parentale d’un chromosome), avec, par exemple, une duplication maternelle et une délétion paternelle. Bruce Cattanach et son équipe établirent ainsi, au cours des vingt dernières années, une carte complète des onze régions chromosomiques non équivalentes selon qu’elles sont héritées du père ou de la mère, et qui affectent la croissance, le comportement ou la viabilité des souris[1] [3, 4]. Ces résultats ont permis de préciser les hypothèses dérivées des premières expériences de transfert nucléaire : certains gènes, essentiels pour le développement, ne sont exprimés qu’à partir d’un seul chromosome, maternel ou paternel, et sont soumis à une empreinte parentale.
Ces expériences pionnières ont été suivies par la mise en évidence des premiers gènes soumis à empreinte vers 1990. Les expériences d’invalidation de gènes, utilisant les cellules ES et la recombinaison homologue, prenaient leur essor à ce moment-là. Deux gènes ont été ainsi invalidés, ceux de l’Igf2 (insulin-like growth factor 2) et de son récepteur Igf2R [5, 6]. De façon surprenante, les croisements réciproques des souris dont ces gènes ont été invalidés ne donnent pas le même résultat : par exemple, si la mutation nulle du gène Igf2 est transmise par la femelle, les descendants sont de taille normale ; en revanche, si la mutation nulle est transmise par le mâle, les souriceaux obtenus sont 40 % plus petits. L’explication vient du fait que le gène Igf2 ne s’exprime qu’à partir de l’allèle d’origine paternelle, car il est soumis à une empreinte parentale. Chez les hétérozygotes portant la mutation nulle d’Igf2 sur le chromosome d’origine paternelle, aucune expression d’Igf2 à partir du chromosome d’origine maternelle ne compense la délétion, et ces souris hétérozygotes ont un phénotype identique à celui de souris homozygotes pour la mutation nulle.
Si la plupart des gènes des mammifères sont exprimés de façon bi-allélique, certaines régions chromosomiques portent des gènes dont l’expression est mono-allélique et dépend de l’origine parentale. Ces gènes, au nombre d’une soixantaine aujourd’hui, sont marqués par une empreinte parentale qui détermine leur expression différentielle aussi bien au cours du développement embryonnaire que pendant la vie adulte.
Caractéristiques de l’empreinte
La plupart des gènes soumis à empreinte sont regroupés en grandes régions chromosomiques (clusters), ce qui suggère l’existence d’éléments capables de contrôler à distance des gènes multiples. Ces domaines, qui peuvent couvrir jusqu’à 4 Mb, comportent des gènes exprimés aussi bien à partir de l’allèle maternel qu’à partir de l’allèle paternel. D’abord identifiées chez la souris, ces régions sont parfaitement conservées chez l’homme, certaines d’entre elles étant associées à des maladies liées à l’origine parentale (voirplus loin).
Méthylation de l’ADN
L’empreinte parentale est caractérisée par une méthylation différentielle des dinucléotides CpG de l’ADN. Des éléments de contrôle, appelés DMR (differentially methylated regions), ont été identifiés au niveau de la plupart des gènes soumis à empreinte ; la méthylation de ces séquences contrôle la transcription des gènes concernés. La première mise en évidence de l’importance de la méthylation dans le mécanisme d’empreinte parentale est venue de l’analyse de souris portant une délétion de l’ADN méthyltransférase 1 (DNMT1) [7]. Cette délétion entraîne une létalité des embryons au jour 9 (E9), preuve de l’importance de la méthylation dans le développement embryonnaire des mammifères. Plus spécifiquement, les embryons DNMT1-/- pris avant E9 ont été analysés pour la méthylation des quelques gènes soumis à empreinte connus à cette époque. Tous ces gènes avaient une expression totalement perturbée : par exemple, le gène H19, normalement exprimé à partir de l’allèle maternel et possédant une région DMR méthylée sur l’allèle paternel, était totalement déméthylé et exprimé de façon bi-allélique. Depuis la DNMT1, d’autres DNMT ont été identifiées : les DNMT3A, -3B et -3L ont pour fonction de méthyler l’ADN de novo (dans les cellules germinales, par exemple), tandis que la DNMT1 est impliquée dans le maintien de la méthylation au cours des divisions cellulaires [8, 9].
L’établissement de la méthylation sur une séquence d’ADN s’accompagne également d’un changement de conformation de la chromatine, en raison de modifications post-traductionnelles des histones ((→) m/s 2005, n° 4, p. 384). Des enzymes capables de modifier ces histones, comme les histone-désacétylases ou les histone-méthyltransférases, participent au processus complexe de remodelage de la chromatine (il s’agit des protéines appartenant au complexe Polycomb, telles que EZH2 et EED) [10, 11].
Ces modifications des histones sont également impliquées dans la régulation des gènes soumis à empreinte, et sont étroitement associées à la méthylation de l’ADN. Il est possible que la modification des histones puisse, à elle seule, constituer une marque épigénétique suffisante pour contrôler l’empreinte. Mais la méthylation de l’ADN intervient probablement dans un second temps pour imposer une marque différentielle qui joue un rôle de verrouillage de la structure chromatinienne [12, 13].
Mise en place de l’empreinte
L’établissement des différences épigénétiques entre les deux chromosomes parentaux a lieu au cours de la gamétogenèse mâle et femelle. Avant cet établissement dans la lignée germinale, les empreintes des parents sont effacées et les nouvelles empreintes sont établies selon le sexe du nouvel embryon (Figure 1A) : cet effacement, qui a lieu dans les cellules germinales primordiales (PGC, primordial germ cells), s’effectue par la perte de méthylation de l’ADN.
Figure 1
L’empreinte parentale.
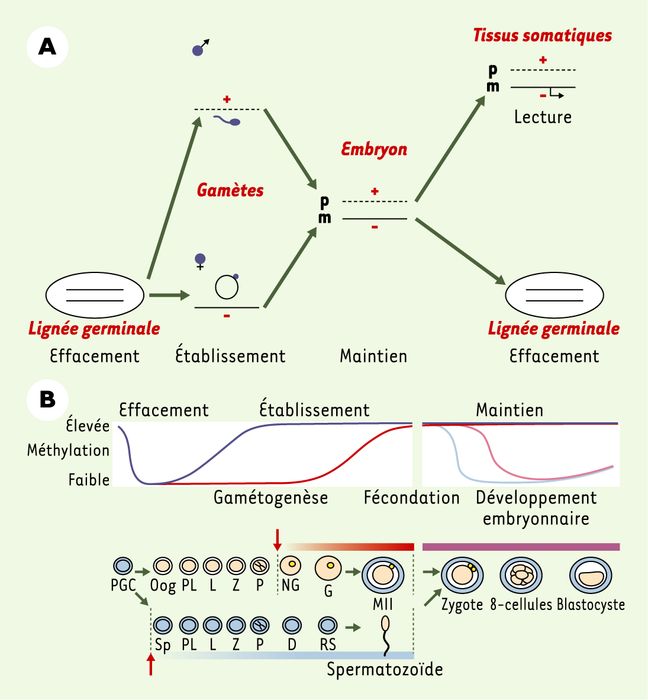
A. Cycle de l’empreinte parentale. Les étapes de la mise en place de l’empreinte parentale sont l’effacement de l’empreinte sur les deux chromosomes parentaux dans la nouvelle lignée germinale, l’établissement d’une nouvelle empreinte au cours de la gamétogenèse, suivant le sexe de l’embryon, le maintien de l’empreinte au cours des divisions cellulaires et, enfin, la lecture de l’empreinte dans les tissus somatiques, se traduisant par une expression mono-allélique des gènes soumis à empreinte. B. Méthylation et déméthylation programmées du génome des ovocytes, des spermatozoïdes et des embryons. En haut : état de méthylation des gènes soumis à l’empreinte maternelle (rouge) et paternelle (bleu). La méthylation des gènes non soumis à empreinte suit ce même profil pendant la gamétogenèse, mais décline après la fécondation (maternels en rose et paternels en bleu clair). En bas : établissement de l’empreinte pendant l’ovogenèse et la spermatogenèse. La méthylation est ensuite maintenue sur les gènes soumis à empreinte dans l’embryon pré-implantatoire. Les deux flèches rouges indiquent le moment de la naissance par rapport à la gamétogenèse (adapté de [29]).
Les PGC sont les cellules précurseurs des cellules germinales. Elles apparaissent dans l’allantoïde de l’embryon de souris au jour 7,5, pour migrer ensuite vers les gonades et les coloniser à partir du jour 10,5. L’analyse de l’état de méthylation de certains gènes soumis à empreinte (Encadré), réalisée par la technique, très sensible, du traitement par le bisulfite suivi d’un séquençage, a montré la perte de méthylation spécifique d’allèle liée à l’empreinte à partir du jour 13,5. On observe donc un processus d’effacement de l’empreinte dans ces PGC, suivi d’une méthylation de novo (phase d’établissement) dans les cellules germinales mâles et femelles [14] (Figure 1B). Le moment précis de cette mise en place de la méthylation a été déterminé dans chaque lignée germinale. Elle intervient chez le mâle dans les gonocytes quiescents (période prénatale), pour être complète au stade pachytène des spermatogonies après la naissance. Chez la femelle, la méthylation n’est complète qu’après la naissance, dans les ovocytes matures [15].
L’importance fonctionnelle de la méthylation différentielle des génomes parentaux a également été démontrée par des expériences de transfert nucléaire, dans lesquelles des embryons parthénogénotes contenant le noyau d’un ovocyte quiescent immature et le noyau d’un ovocyte mature sont capables de survivre trois jours de plus (E13,5) qu’un même embryon contenant deux noyaux provenant de deux ovocytes matures [16] : cela est dû à la différence d’empreinte parentale dans ces deux noyaux, l’empreinte étant complètement établie dans le noyau mature, mais pas encore dans le noyau immature. Très récemment, Tomohiro Kono a utilisé comme source de noyau immature des ovocytes de souris dont le gène H19 a été invalidé ; ce gène produit un ARN non codant dont la fonction est inconnue, mais sa délétion entraîne la surexpression du gène Igf2 adjacent. Les souris obtenues sont viables, ce qui montre qu’une parthénogenèse expérimentale est possible chez la souris. Il reste cependant à comprendre comment la délétion du gène H19 peut rétablir une différence fonctionnelle entre deux génomes de même origine parentale [17].
Méthylation et développement embryonnaire
Après la fécondation, les premières phases du développement de l’embryon sont caractérisées par une vague de déméthylation complète du génome, suivie, avant l’implantation, d’une reméthylation des gènes zygotiques au stade blastocyste. Cette déméthylation massive aurait pour rôle d’effacer toute régulation provenant des cellules germinales parentales pour permettre un nouveau profil d’expression génique caractéristique du zygote. Il est frappant de constater que les méthylations spécifiques des gènes soumis à empreinte, établies pendant la gamétogenèse, sont complètement protégées de cette déméthylation, sans que l’on comprenne encore le mécanisme de cette protection.
Contrôle de l’empreinte en cis et en trans
Certaines DMR (differentially methylated regions), cibles de la méthylation différentielle, ont été définies comme centres d’empreinte (IC, imprinting center). Ces séquences sont souvent considérées comme des DMR primaires, dont la méthylation a lieu dans les gamètes, par opposition aux DMR secondaires, dont la méthylation différentielle est plus aléatoire et peut avoir lieu après la fécondation. Ces IC sont les éléments régulateurs capables de contrôler en cis l’expression mono-allélique de l’ensemble des gènes d’un domaine [18].
Des facteurs agissant en trans ont également été identifiés. Associées aux méthyltransférases de l’ADN et aux protéines de modification des histones, on trouve les MBD (methyl CpG binding proteins), ainsi que des protéines insulatrices, capables de constituer des barrières au sein d’une unité transcriptionnelle, comme le facteur CTCF (CCCTC-binding factor)[19]. Celui-ci, par exemple, se fixe sur l’IC du locus H19-Igf2 et joue un rôle d’insulateur par rapport à un enhancer commun à ces deux gènes, expliquant le mécanisme de compétition des enhancers décrit pour ce locus (Figure 2A).
Figure 2
Régulation en cluster de l’expression mono-allélique des gènes soumis à empreinte.

La région distale du chromosome 7 murin comporte deux domaines et deux centres d’empreinte indépendants (IC1 et IC2). A. Domaine 1 : les enhancers situés en aval du gène H19 interagissent avec le promoteur maternel du gène H19, mais ne peuvent pas activer l’allèle maternel du gène Igf2 en raison de la frontière produite par la protéine insulatrice CTCF, fixée sur la DMR maternelle (IC1) non méthylée. La DMR paternelle hyperméthylée réprime quant à elle l’allèle paternel du gène H19, en agissant comme un silencer [28]. B. Le domaine 2 comporte une dizaine de gènes, dont certains sont soumis à empreinte seulement dans le placenta chez la souris (Nap1l4, Tssc4, CD81, Ascl2). L’IC2 (ou KvDMR), localisée dans l’intron 10 de KvLQT1, est une DMR méthylée sur l’allèle maternel et non méthylée sur l’allèle paternel. LIT1 (LQT intronic transcript 1) est un ARN non codant antisens du gène KvLQT1, exprimé à partir de la DMR paternelle, et qui permettrait la répression paternelle des gènes Phlda2, Tssc5, p57kip2, KvLQT1. C. Locus Igf2R : dans l’intron 2 du gène Igf2R se trouve le centre d’empreinte (ICE, imprinting control element), qui est une DMR méthylée sur l’allèle maternel. À partir de ce promoteur paternel non méthylé s’exprime l’ARN antisens AIR, qui serait responsable de la répression en cis des gènes Igf2R, Slc22a2 et Slc22a3. D. Locus 7C murin ou PWS-AS humain : le centre d’empreinte du locus PWS-AS comporte deux modules, AS-SRO et PWS-SRO (IC). Ce dernier est une DMR méthylée sur l’allèle maternel. Par son action en cis, elle permettrait d’activer l’expression des gènes MRKN3, NDN et MAGEL2, et de réprimer le gène UBE3A, grâce à l’expression de l’ARN non codant UBE3A-ATS.
De plus, les produits de certains gènes soumis à empreinte sont des ARN non codants dont le rôle n’est pas encore bien défini. Ces ARN pourraient intervenir par une fonction antisens ou par un mécanisme d’interférence par l’ARN (ARNsi ou microARN) dans la régulation de l’expression des gènes adjacents.
Nature de l’empreinte
Il reste à identifier la nature même de l’empreinte ; on peut supposer qu’il existe un complexe d’initiation de l’empreinte, capable de reconnaître des séquences spécifiques, telles que les centres d’empreinte, et que ce complexe ne peut être présent que dans la lignée germinale. La propagation ultérieure de l’empreinte peut se faire à l’aide des MBD, des DNMT et des protéines de remodelage de la chromatine. Cette empreinte, qui se traduit par une méthylation différentielle, est ensuite capable de moduler l’expression mono-allélique des gènes dans les cellules somatiques de façon temporelle et dépendante du tissu. Le complexe d’initiation et les facteurs additionnels qui permettent d’identifier les séquences cibles de l’empreinte dans la lignée germinale sont encore inconnus.
Exemples de régulation des régions soumises à l’empreinte
Comme cela a été évoqué précédemment, les gènes soumis à empreinte sont regroupés dans le génome en domaines chromosomiques. Afin d’expliquer le contrôle à distance observé pour ces gènes, deux mécanismes de régulation sont proposés : d’une part, les centres d’empreinte, différentiellement méthylés, sont capables de contrôler l’expression mono-allélique maternelle ou paternelle de l’ensemble des gènes d’un domaine ; d’autre part, plus d’un quart de ces gènes produisent des ARN non codants ; certains d’entre eux jouent le rôle d’antisens capables d’intervenir dans la régulation de l’expression des gènes adjacents. Trois exemples pemettent d’illustrer les différents aspects de cette régulation.
Chez la souris, dans la partie distale du chromosome 7, une région chromosomique couvrant 1 Mb est constituée de deux domaines possédant chacun leur propre centre d’empreinte indépendant, appelé IC1 et IC2. Le domaine 1 (Figure 2A) comporte les gènes Igf2 et H19. La protéine CTCF se fixe sur l’IC1 (également appelé DMR H19) non méthylé de l’allèle maternel, pour constituer une frontière, comme cela est décrit ci-dessus. Le domaine 2 contient l’IC2, une DMR située dans un intron du gène KvLQT1 (Figure 2B). Cette DMR, non méthylée sur l’allèle paternel, sert de promoteur à un ARN antisens LIT1 de 80 kb, transcrit à partir de cet allèle et en partie complémentaire du gène KvLQT1 [20].
Un mécanisme analogue a été décrit sur le locus Igf2R (Figure 2C) : l’expression paternelle, à partir de l’ICE, d’un grand ARN antisens de plus de 100 kb, appelé AIR (antisense of Igf2R), inhibe en cis l’expression des gènes Igf2R, Slc22a2 et Slc22a3 [21, 22].
Enfin, une autre région chromosomique de 4 Mb (dans la région centrale du chromosome 7, ou 7C) contient les gènes Snrpn, Necdin (Ndn), Mrkn3 (Zfp127) et Magel2, exprimés exclusivement à partir de l’allèle paternel, et le gène Ube3a, à expression maternelle dans le cerveau uniquement (Figure 2D). Le centre d’empreinte du locus comporte une DMR méthylée sur l’allèle maternel (PWS-SRO) au niveau du promoteur Snrpn. Un très grand ARN non codant d’environ 1 000 kb, appelé Ube3a-Ats (Ube3a antisense), exprimé à partir de l’allèle paternel, est complémentaire des gènes Snrpn et Ube3a. Le centre d’empreinte contrôlerait en cis à la fois l’expression paternelle des gènes en amont de ce centre et, par l’intermédiaire de l’ARN antisens Ube3a-Ats, la répression paternelle d’Ube3a [23, 24].
Le mécanisme d’action de ces ARN antisens reste encore à être défini, mais il semble qu’il fait intervenir la compaction de la chromatine et pourrait également impliquer la méthylation de l’ADN [25, 26].
Empreinte parentale et maladies
Ces exemples suggèrent que l’empreinte parentale est contrôlée par différents mécanismes étroitement corrélés. Leur dérèglement peut perturber l’équilibre d’expression de multiples gènes d’une région, conduisant à l’apparition de plusieurs maladies humaines associées à ces domaines d’empreinte [27].
Ainsi, la région 11p15 chez l’homme, synthénique de la partie distale du chromosome 7 murin, est associée au syndrome de Beckwith-Wiedemann. Celui-ci se caractérise par un ensemble de malformations congénitales (liées au domaine 1) et une prédisposition à certaines tumeurs (probablement liée au domaine 2).
Une autre région très étudiée chez l’homme est le locus 15q11-q13, synthénique du locus 7C murin, associé aux syndromes de Prader-Willi (PWS) et d’Angelman (AS). Ces maladies sont caractérisées essentiellement par des retards mentaux. Le PWS serait causé par la perte d’expression des gènes SNRPN, etc., par unidisomie maternelle ou délétion paternelle. L’AS serait dû, quant à lui, à l’absence de protéine UBE3A fonctionnelle par unidisomie paternelle, délétion maternelle ou mutation.
Ces maladies sont donc associées soit à des pertes d’hétérozygotie (LOH, loss of heterozygocity), ayant pour résultat la duplication maternelle ou paternelle de la région, ce qui entraîne la surexpression de certains gènes et l’extinction complète d’autres gènes, soit à des pertes d’empreinte (LOI, loss of imprinting), qui affectent particulièrement les centres d’empreinte, montrant ainsi leur importance dans la régulation coordonnée de plusieurs gènes d’un même domaine soumis à empreinte.
Conclusions
L’empreinte parentale est un mécanisme épigénétique de contrôle transcriptionnel, dont l’étude s‘est beaucoup développée au cours de ces vingt dernières années. Les différents aspects de cette régulation ont été élucidés, avant tout, grâce à des études réalisées in vivo sur des modèles de souris. La méthylation de l’ADN, ainsi que les modifications post-traductionnelles des histones, participent à l’établissement de l’expression mono-allélique des gènes soumis à empreinte. Il reste encore à identifier la nature même de l’empreinte, c’est-à-dire la façon dont la machinerie chromatinienne est capable de choisir et de reconnaître les cibles de ce marquage différentiel. L’identification de l’empreinte parentale et des mécanismes moléculaires qui l’accompagnent devrait permettre d’envisager une meilleure compréhension des maladies liées à des modifications épigénétiques.
Parties annexes
Note
Références
- 1. McGrath J, Solter D. Completion of embryogenesis requires both the maternal and paternal genomes. Cell 1984 ; 37 : 179-83.
- 2. Surani MA, Barton S, Norris M. Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature 1984 ; 308 : 548-50.
- 3. Cattanach BM, Kirk M. Differential activity of maternally and paternally derived chromosome regions in mice. Nature 1985 ; 315 : 496-8.
- 4. Cattanach BM, Beechey CV, Peters J. Interactions between imprinting effects in the mouse. Genetics 2004 ; 168 : 397-413.
- 5. DeChiara TM, Efstratiadis A, Robertson EJ. A growth deficiency phenotype in heterozygous mice carrying an insulin-like growth factor II gene disrupted by gene targeting. Nature 1990 ; 345 : 78-80.
- 6. Barlow DP, Stoger R, Hermann BG, et al. The mouse insulin-like growth factor type 2 receptor is imprinted and closely linked to the Tme locus. Nature 1991 ; 349 : 84-7.
- 7. Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 1992 ; 69 : 915-26.
- 8. Hata K, Okano M, Lei H, Li E. Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development 2002 ; 129 : 1983-93.
- 9. Bourc’his D, Xu GL, Lin CS, et al. Dnmt3L and the establishment of maternal genomic imprints. Science 2001 ; 294 : 2536-9.
- 10. Li E. Chromatin modification and epigenetic reprogramming in mammalian development. Nat Rev Genet 2002 ; 3 : 662-73.
- 11. Mager J, Montgomery ND, de Villena FP, Magnuson T. Genome imprinting regulated by the mouse Polycomb group protein Eed. Nat Genet 2003 ; 33 : 502-7.
- 12. Umlauf D, Goto Y, Cao R, et al. Imprinting along the Kcnq1 domain on mouse chromosome 7 involves repressive histone methylation and recruitment of Polycomb group complexes. Nat Genet 2004 ; 36 : 1296-300.
- 13. Lewis A, Mitsuya K, Umlauf D, et al. Imprinting on distal chromosome 7 in the placenta involves repressive histone methylation independent of DNA methylation. Nat Genet 2004 ; 36 : 1291-5.
- 14. Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science 2001 ; 293 : 1089-93.
- 15. Lucifero D, Mann MR, Bartolomei MS, Trasler JM. Gene-specific timing and epigenetic memory in oocyte imprinting. Hum Mol Genet 2004 ; 13 : 839-49.
- 16. Obata Y, Kaneko-Ishino T, Koide T, et al. Disruption of primary imprinting during oocyte growth leads to the modified expression of imprinted genes during embryogenesis. Development 1998 ; 125 : 1553-60.
- 17. Kono T, Obata Y, Wu Q, et al. Birth of parthenogenetic mice that can develop to adulthood. Nature 2004 ; 428 : 860-4.
- 18. Lopes S, Lewis A, Hajkova P, et al. Epigenetic modifications in an imprinting cluster are controlled by a hierarchy of DMRs suggesting long-range chromatin interactions. Hum Mol Genet 2003 ; 12 : 295-305.
- 19. Hark AT, Schoenherr CJ, Katz DJ, et al. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 2000 ; 405 : 486-9.
- 20. Lee MP, DeBaun MR, Mitsuya K, et al. Loss of imprinting of a paternally expressed transcript, with antisense orientation to KVLQT1, occurs frequently in Beckwith-Wiedemann syndrome and is independent of insulin-like growth factor II imprinting. Proc Natl Acad Sci USA 1999 ; 96 : 5203-8.
- 21. Wutz A, Smrzka OW, Schweifer N, et al. Imprinted expression of the Igf2r gene depends on an intronic CpG island. Nature 1997 ; 389 : 745-9.
- 22. Sleutels F, Zwart R, Barlow DP. The non-coding Air RNA is required for silencing autosomal imprinted genes. Nature 2002 ; 415 : 810-3.
- 23. Rougeulle C, Cardoso C, Fontes M, et al. An imprinted antisense RNA overlaps UBE3A and a second maternally expressed transcript. Nat Genet 1998 ; 19 : 15-6.
- 24. Landers M, Bancescu DL, Le Meur E, et al. Regulation of the large (approximately 1000 kb) imprinted murine Ube3a antisense transcript by alternative exons upstream of Snurf/Snrpn. Nucleic Acids Res 2004 ; 32 : 3480-92.
- 25. Rougeulle C, Heard E. Antisense RNA in imprinting: spreading silence through Air. Trends Genet 2002 ; 18 : 434-7.
- 26. Thakur N, Tiwari VK, Thomassin H, et al. An antisense RNA regulates the bidirectional silencing property of the Kcnq1 imprinting control region. Mol Cell Biol 2004 ; 24 : 7855-62.
- 27. Verona RI, Mann MR, Bartolomei MS. Genomic imprinting: intricacies of epigenetic regulation in clusters. Annu Rev Cell Dev Biol 2003 ; 19 : 237-59.
- 28. Drewell RA, Brenton JD, Ainscough JF, et al. Deletion of a silencer element disrupts H19 imprinting independently of a DNA methylation epigenetic switch. Development 2000 ; 127 : 3419-28.
- 29. Gosden R, Trasler J, Lucifero D, Faddy M. Rare congenital disorders, imprinted genes, and assisted reproductive technology. Lancet 2003 ; 361 : 1975-7.
Liste des figures
Figure 1
L’empreinte parentale.
A. Cycle de l’empreinte parentale. Les étapes de la mise en place de l’empreinte parentale sont l’effacement de l’empreinte sur les deux chromosomes parentaux dans la nouvelle lignée germinale, l’établissement d’une nouvelle empreinte au cours de la gamétogenèse, suivant le sexe de l’embryon, le maintien de l’empreinte au cours des divisions cellulaires et, enfin, la lecture de l’empreinte dans les tissus somatiques, se traduisant par une expression mono-allélique des gènes soumis à empreinte. B. Méthylation et déméthylation programmées du génome des ovocytes, des spermatozoïdes et des embryons. En haut : état de méthylation des gènes soumis à l’empreinte maternelle (rouge) et paternelle (bleu). La méthylation des gènes non soumis à empreinte suit ce même profil pendant la gamétogenèse, mais décline après la fécondation (maternels en rose et paternels en bleu clair). En bas : établissement de l’empreinte pendant l’ovogenèse et la spermatogenèse. La méthylation est ensuite maintenue sur les gènes soumis à empreinte dans l’embryon pré-implantatoire. Les deux flèches rouges indiquent le moment de la naissance par rapport à la gamétogenèse (adapté de [29]).
Figure 2
Régulation en cluster de l’expression mono-allélique des gènes soumis à empreinte.
La région distale du chromosome 7 murin comporte deux domaines et deux centres d’empreinte indépendants (IC1 et IC2). A. Domaine 1 : les enhancers situés en aval du gène H19 interagissent avec le promoteur maternel du gène H19, mais ne peuvent pas activer l’allèle maternel du gène Igf2 en raison de la frontière produite par la protéine insulatrice CTCF, fixée sur la DMR maternelle (IC1) non méthylée. La DMR paternelle hyperméthylée réprime quant à elle l’allèle paternel du gène H19, en agissant comme un silencer [28]. B. Le domaine 2 comporte une dizaine de gènes, dont certains sont soumis à empreinte seulement dans le placenta chez la souris (Nap1l4, Tssc4, CD81, Ascl2). L’IC2 (ou KvDMR), localisée dans l’intron 10 de KvLQT1, est une DMR méthylée sur l’allèle maternel et non méthylée sur l’allèle paternel. LIT1 (LQT intronic transcript 1) est un ARN non codant antisens du gène KvLQT1, exprimé à partir de la DMR paternelle, et qui permettrait la répression paternelle des gènes Phlda2, Tssc5, p57kip2, KvLQT1. C. Locus Igf2R : dans l’intron 2 du gène Igf2R se trouve le centre d’empreinte (ICE, imprinting control element), qui est une DMR méthylée sur l’allèle maternel. À partir de ce promoteur paternel non méthylé s’exprime l’ARN antisens AIR, qui serait responsable de la répression en cis des gènes Igf2R, Slc22a2 et Slc22a3. D. Locus 7C murin ou PWS-AS humain : le centre d’empreinte du locus PWS-AS comporte deux modules, AS-SRO et PWS-SRO (IC). Ce dernier est une DMR méthylée sur l’allèle maternel. Par son action en cis, elle permettrait d’activer l’expression des gènes MRKN3, NDN et MAGEL2, et de réprimer le gène UBE3A, grâce à l’expression de l’ARN non codant UBE3A-ATS.