Résumés
Résumé
L’architecture et la fonction des fibres myélinisées sont dépendantes de l’établissement de contacts cellulaires finement réglés entre les membranes d’une même cellule gliale myélinisante, entre l’axone et les cellules gliales, et entre les cellules gliales et la matrice extracellulaire. Des composants protéiques majeurs de l’ensemble de ces contacts ont été identifiés ces dernières années. Des progrès importants ont notamment été faits dans l’identification de complexes moléculaires impliqués dans les contacts axo-gliaux au niveau des noeuds de Ranvier. Le rôle capital de certains composants des contacts dans le maintien de l’intégrité structurale et fonctionnelle des fibres a été démontré par la production de modèles murins. Dans certains cas, des mutations des gènes correspondants ont été identifiées chez des patients atteints de neuropathies périphériques telles que les maladies de Charcot-Marie-Tooth (CMT).
Summary
Myelination allows the fast propagation of action potentials at a low energetic cost. It provides an insulating myelin sheath regularly interrupted at nodes of Ranvier where voltage-gated Na+ channels are concentrated. In the peripheral nervous system, the normal function of myelinated fibers requires the formation of highly differentiated and organized contacts between the myelinating Schwann cells, the axons and the extracellular matrix. Some of the major molecular complexes that underlie these contacts have been identified. Compact myelin which forms the bulk of the myelin sheath results from the fusion of the Schwann cell membranes through the proteins P0, PMP22 and MBP. The basal lamina of myelinating Schwann cells contains laminin-2 which associates with the glial complex dystroglycan/DPR2/L-periaxin. Non compact myelin, found in paranodal loops, periaxonal and abaxonal regions, and Schmidt-Lanterman incisures, presents reflexive adherens junctions, tight junctions and gap junctions, which contain cadherins, claudins and connexins, repectively. Axo-glial contacts determine the formation of distinct domains on the axon, the node, the paranode, and the juxtaparanode. At the paranodes, the glial membrane is tightly attached to the axolemma by septate-like junctions. Paranodal and juxtaparanodal axoglial complexes comprise an axonal transmembrane protein of the NCP family associated in cis and in trans with cell adhesion molecules of the immunoglobulin superfamily (IgSF-CAM). At nodes, axonal complexes are composed of Na+ channels and IgSF-CAMs. Schwann cell microvilli, which loosely cover the node, contain ERM proteins and the proteoglycans syndecan-3 and -4. The fundamental role of the cellular contacts in the normal function of myelinated fibers has been supported by rodent models and the detection of genetic alterations in patients with peripheral demyelinating neuropathies such as Charcot-Marie-Tooth diseases. Understanding more precisely their molecular basis now appears essential as a requisite step to further examine their involvement in the pathogenesis of peripheral neuropathies in general.
Corps de l’article
Dans le système nerveux, les signaux sont transmis sur de longues distances par les neurones qui produisent et conduisent le long de leur axone des potentiels d’action. Ces potentiels d’action proviennent d’un flux d’ions à travers des canaux Na+ dépendants du voltage, qui modifie la distribution des charges de part et d’autre de la membrane (Figure 1). Ce mécanisme de base est similaire dans tous les neurones.
Figure 1
Conduction de l’influx nerveux dans les fibres myélinisées et non myélinisées.

Les potentiels d’action transmis le long des axones sont produits par un flux d’ions à travers des canaux Na+ dépendants du voltage, qui modifie la distribution des charges de part et d’autre de la membrane. A. Dans les fibres non myélinisées, les canaux Na+ sont répartis de façon uniforme dans la membrane axonale, et le potentiel d’action est transmis de façon continue le long de l’axone à une vitesse ne dépassant pas 3 m/s. B. Dans les fibres myélinisées, les canaux Na+ sont concentrés dans des régions discontinues de l’axone, appelées noeuds de Ranvier, permettant une propagation rapide, dite « saltatoire », du potentiel d’action par bonds d’un noeud à l’autre. Les noeuds de Ranvier sont séparés par une gaine de myéline isolante, formée par les cellules de Schwann dans le système nerveux périphérique, et par les oligodendrocytes dans le système nerveux central.
La vitesse de conduction est un paramètre fonctionnellement très important. Deux mécanismes distincts ont évolué en parallèle pour augmenter cette vitesse. L’augmentation de la rapidité de propagation des potentiels d’action peut être atteinte en augmentant le diamètre de l’axone. Ce mécanisme facilite efficacement la conduction chez les invertébrés. Un autre mécanisme de conduction rapide s’est mis en place chez les vertébrés à mâchoire. Une gaine de myéline recouvre et isole des segments d’axone, et concentre les canaux Na+ dépendants du voltage dans des régions discontinues de l’axone, appelées noeuds de Ranvier (Figure 1). Ainsi, l’isolement par segments de l’axone et la concentration des canaux Na+ au niveau des noeuds de Ranvier contribuent à une propagation rapide du potentiel d’action, par bonds d’un noeud à l’autre. Cette propagation, dite « saltatoire », équivaut sur un plan fonctionnel à augmenter le diamètre de la membrane axonale jusqu’à 100 fois. La gaine de myéline est formée par des cellules gliales myélinisantes qui s’enroulent autour de l’axone : cellules de Schwann dans le système nerveux périphérique (SNP) et oligodendrocytes dans le système nerveux central (SNC).
Dans le SNP, l’architecture et la fonction des fibres myélinisées reposent sur l’établissement d’interactions cellulaires finement réglées, d’une part entre l’axone et les cellules gliales myélinisantes (interactions hétérotypiques axo-gliales) et, d’autre part, entre les membranes d’une même cellule gliale myélinisante (interactions autotypiques). Ces interactions résultent de la mise en place de différents types de contacts impliquant des protéines transmembranaires et d’adhérence, des protéines d’échafaudage et des éléments du cytosquelette. L’intégrité des fibres myélinisées dans le SNP nécessite également la mise en place et le maintien d’interactions entre la cellule gliale myélinisante et la matrice extracellulaire. Les cellules de Schwann myélinisantes établissent notamment des interactions étroites avec la lame basale qui les entoure. L’importance fonctionnelle de toutes ces interactions autotypiques, axo-gliales ou cellule/matrice a été particulièrement soulignée ces dernières années par la production de souris dont les gènes codant pour certaines des protéines impliquées dans ces interactions ont été invalidés (Tableau I). Certaines neuropathies périphériques ont par ailleurs été associées à la présence de mutations sur les gènes codant pour des protéines des contacts cellulaires (Tableau I). Parmi ces neuropathies, les plus fréquemment représentées sont les maladies de Charcot-Marie-Tooth (CMT), neuropathies sensorielles et motrices très hétérogènes dans lesquelles on distingue des formes démyélinisantes et des formes axonales.
Tableau I
Importance fonctionnelle des contacts cellulaires dans les fibres myélinisées du système nerveux périphérique.
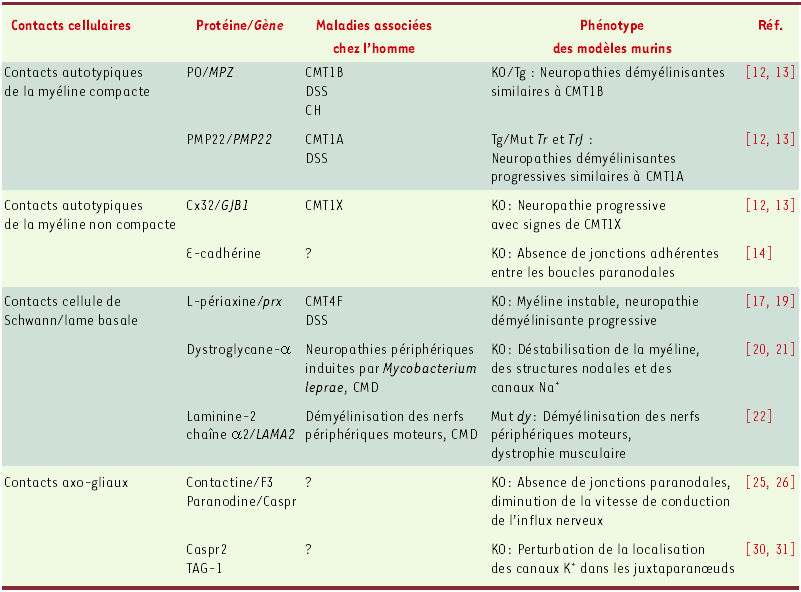
CMT : maladie de Charcot-Marie-Tooth ; DSS : neuropathie récessive de Dejerine-Sottas ; CH : hypomyélinisation congénitale ; CMD : dystrophie musculaire congénitale ; KO : souris dont les gènes ont été invalidés ; Tg : souris transgéniques ; Mut : souris présentant des mutations spontanées ; PMP22 : peripheral myelin protein 22 ; PO : myelin protein zero ; Cx : connexine ; Caspr : contactin-associated protein ; TAG-1 : transiently-expressed axonal glycoprotein 1.
Cellules gliales myélinisantes et interactions autotypiques
La gaine de myéline peut être subdivisée en deux sous-domaines structurellement et biochimiquement distincts : la myéline compacte et la myéline non compacte.
La myéline compacte est localisée dans les internoeuds et correspond à des régions où la cellule gliale myélinisante est dépourvue de cytoplasme (Figure 2). Elle est essentiellement constituée de lipides (70 %) et résulte de l’accolement des membranes adjacentes de la cellule de Schwann lors de son enroulement progressif autour de l’axone par un mécanisme impliquant la formation de tétramères d’une protéine transmembranaire, la protéine P0 (myelin protein zero) (Figure 3A) [1]. Cette protéine joue le rôle de molécule d’adhérence grâce à la capacité de son domaine extracellulaire de type « immunoglobuline » à établir des interactions homotypiques en cis et en trans [2]. La protéine P0 représente plus de 50 % des protéines de la myéline périphérique. Deux autres protéines majeures de cette myéline sont la protéine MBP (myelin basic protein) et PMP22 (peripheral myelin protein 22), une protéine intégrale comportant quatre domaines transmembranaires (Figure 3A) [1].
Figure 2
Organisation ultrastructurale d’une fibre myélinisée du système nerveux périphérique.
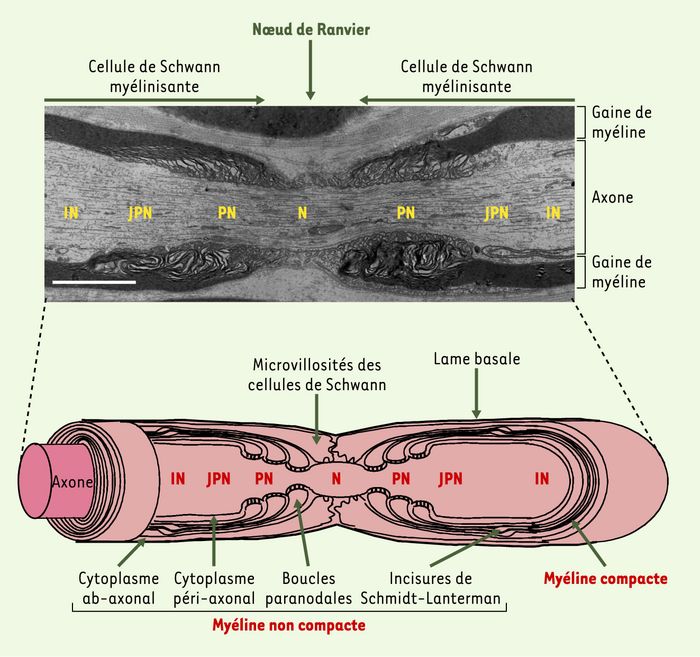
La myéline se divise en deux sous-domaines structurellement et fonctionnement distincts : la myéline compacte et la myéline non compacte. La myéline compacte est localisée dans les internoeuds (IN) situés entre les noeuds de Ranvier, et résulte de l’accolement des membranes adjacentes de la cellule de Schwann lors de son enroulement progressif autour de l’axone. Dans ces régions, la myéline ne contient plus de cytoplasme. La myéline non compacte correspond, quant à elle, à des régions de la cellule de Schwann où persiste du cytoplasme. Ces régions correspondent aux boucles gliales paranodales, aux incisures de Schmidt-Lanterman, aux régions péri-axonale et ab-axonale et aux microvillosités. Au cours du développement, l’enroulement de la cellule de Schwann autour de l’axone conduit à la formation de trois types de contacts entre la cellule de Schwann et l’axone, qui définissent trois domaines anatomiquement et fonctionnellement distincts le long de l’axone : le noeud (N), le paranoeud (PN) et le juxtaparanoeud (JPN). Le noeud est recouvert par les microvillosités des cellules de Schwann. Au niveau des paranoeuds, l’axolemme est étroitement associé aux boucles latérales des cellules gliales myélinisantes par des jonctions de type « septé ». Le juxtaparanoeud correspond à une région intermédiaire entre le paranoeud et l’internoeud. Partie supérieure de la figure : photo en microscopie électronique d’un noeud de Ranvier d’une fibre myélinisée de nerf phrénique (échelle, 2 μm).
Figure 3
Organisation moléculaire des interactions gliales autotypiques et de la connexion ente la cellule gliale et la lame basale, dans le système nerveux périphérique.
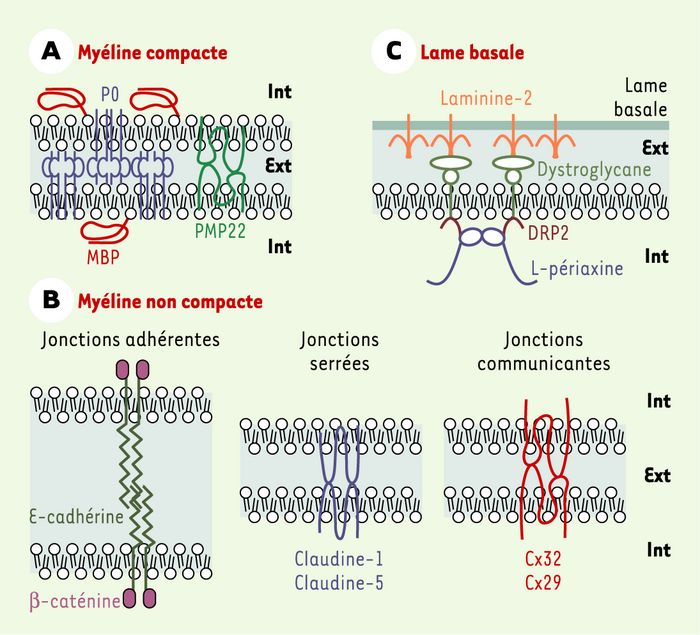
A. Protéines majeures de la myéline compacte. Les trois protéines majeures de la myéline compacte sont les protéines P0 (myelin protein zero), PMP22 (peripheral myelin protein 22) et MBP (myelin basic protein). La protéine P0 joue le rôle de molécule d’adhérence grâce à la capacité de son domaine extracellulaire de type « immunoglobuline » d’établir des interactions homotypiques en cis et en trans. B. Protéines impliquées dans les jonctions autotypiques de la myéline non compacte. Dans les boucles paranodales, les incisures de Schmidt-Lanterman et les régions péri-axonale et ab-axonale, les membranes plasmiques des cellules de Schwann myélinisantes établissent des jonctions autotypiques dites « adhérentes » et « serrées ». Dans les boucles paranodales et les incisures se situent également des jonctions dites « communicantes ». Les jonctions adhérentes contiennent une molécule d’adhérence dépendante du calcium, la E-cadhérine, ainsi que la β-caténine cytoplasmique qui connecte la E-cadhérine aux filaments d’actine. Les protéines identifiées dans les jonctions serrées sont des protéines membranaires intégrales, la claudine-1 et la claudine-5. Les jonctions communicantes se forment par la fusion de deux hémicanaux correspondant à un assemblage de 6 connexines identiques, la connexine 32 (Cx32) ou la connexine 29 (Cx29). C. Protéines impliquées dans la connexion entre la cellule de Schwann myélinisante et sa lame basale. La laminine-2 constitue l’un des composants majeurs de la lame basale. L’un de ses récepteurs sur la cellule de Schwann est le dystroglycane, capable de former des complexes avec deux protéines cytoplasmiques de la cellule de Schwann, DRP2 (dystrophin-related protein 2) et la L-périaxine. Int et Ext : intérieur et extérieur de la cellule de Schwann.
La myéline non compacte correspond à des zones de la cellule gliale myélinisante où persiste du cytoplasme. C’est le cas des boucles dites paranodales, des incisures de Schmidt-Lanterman, des microvillosités nodales et des régions péri-axonale et ab-axonale (Figure 2). Les boucles paranodales sont formées par les bords latéraux de la gaine de myéline au niveau desquels chaque lamelle de myéline compacte s’ouvre en une boucle cytoplasmique. Les incisures de Schmidt-Lanterman sont des régions dans lesquelles les faces cytoplasmiques des membranes gliales ne sont pas adhérentes. Ces régions persistent lors de l’enroulement de la cellule gliale autour de l’axone, formant ainsi une voie cytoplasmique de diffusion qui suit un enroulement hélicoïdal et connecte la région péri-axonale de la cellule de Schwann à sa région ab-axonale [1]. Les microvillosités nodales constituent un prolongement de la cellule de Schwann recouvrant l’axone au niveau du noeud de Ranvier.
Dans les boucles paranodales, les incisures et les régions péri-axonale et ab-axonale, les membranes plasmiques des cellules de Schwann myélinisantes établissent des jonctions autotypiques « adhérentes » et « serrées » (Figure 2) [3, 4]. Les jonctions adhérentes contiennent, comme dans d’autres types cellulaires, une molécule d’adhérence dépendante du calcium, la E-cadhérine, ainsi que la β-caténine cytoplasmique qui connecte la E-cadhérine aux filaments d’actine (Figure 3B) [4]. Les jonctions serrées forment une barrière de perméabilité sélective et réduisent le mouvement des lipides et des protéines dans la membrane plasmique, favorisant ainsi le maintien des régions membranaires spécialisées dans les cellules épithéliales et endothéliales. Il a été proposé que, dans les cellules gliales myélinisantes, ces jonctions entre les membranes adjacentes augmentent leur cohésion et séparent la membrane gliale non compacte de la membrane de la myéline compacte [5]. À l’heure actuelle, les protéines identifiées dans ces jonctions appartiennent à la famille des claudines, protéines membranaires intégrales à 4 domaines transmembranaires [6]. La claudine-1 est localisée dans les boucles paranodales et la région péri-axonale, et la claudine-5 dans les incisures (Figure 3B) [7]. Au niveau des boucles paranodales et des incisures se situent également des jonctions dites « communicantes » [3], formées par la fusion de deux hémicanaux (connexons), chaque connexon correspondant à un assemblage de 6 connexines [8]. Elles facilitent le passage d’ions et de petites molécules entre le cytoplasme ab-axonal et péri-axonal de la cellule de Schwann [9]. Deux membres de la famille des connexines, la connexine 32 (Cx32) et la connexine 29 (Cx29), sont exprimés dans les cellules gliales myélinisantes (Figure 3B) [10, 11].
Des études génétiques récentes soulignent l’importance capitale des protéines PMP22, P0 et Cx32 dans le maintien de l’intégrité des fibres myélinisées périphériques (pour revue, voir [12]). Il apparaît en effet que les altérations du gène PMP22 codant pour la protéine PMP22 constituent aujourd’hui la cause majeure des neuropathies démyélinisantes héréditaires (70 % des cas) [13], le type de neuropathie et la sévérité dépendant du type d’altération génétique (délétion, duplication ou mutation du gène). Le deuxième type d’altérations génétiques le plus fréquemment retrouvé chez les patients atteints de CMT correspond à des mutations du gène GJB1 codant pour la Cx32. On connaît aujourd’hui plus de 240 mutations différentes affectant à la fois les régions codantes et non codantes du gène. Par ailleurs, approximativement 80 mutations différentes du gène MPZ, codant pour la protéine P0, ont été identifiées, toutes associées à des neuropathies périphériques, mais avec des phénotypes variables.
De leur côté, les observations réalisées dans de nombreux modèles animaux confortent ce rôle pathogénique des mutations des protéines Cx32, PMP22 et P0 (pour revue, voir [13]). Des souris dont le gène codant pour la protéine Cx32 a été invalidé développent ainsi une neuropathie progressive avec des signes similaires à ceux d’une forme de CMT (CMT1X). Des souris dont le gène codant pour la protéine P0 a été invalidé présentent une hypomyélinisation sévère ; en revanche, des souris transgéniques surexprimant la protéine P0 sont atteintes de neuropathies démyélinisantes. Enfin, les rats et les souris transgéniques surexprimant de façon modérée le gène PMP22, sous le contrôle de son propre promoteur, développent des signes cliniques semblables à ceux présentés par les patients portant une copie supplémentaire du gène (CMT1A). Une forte surexpression du gène chez les mêmes animaux conduit à une démyélinisation quasi complète des nerfs périphériques.
Le rôle des protéines des jonctions adhérentes dans l’architecture des fibres myélinisées a été souligné par l’invalidation du gène codant pour la E-cadhérine chez la souris [14]. Chez ces souris, la myéline se forme correctement, mais les jonctions adhérentes sont absentes au niveau de la myéline non compacte, notamment entre les boucles paranodales.
Connexion de la cellule de Schwann à sa lame basale
L’un des constituants majeurs de la lame basale entourant la cellule de Schwann myélinisante dans le SNP est la laminine-2, une protéine trimérique pour laquelle la cellule de Schwann possède plusieurs récepteurs, dont le dystroglycane (Figure 3C) [15]. Le dystroglycane est une molécule dimérique constituée d’une sous-unité α (dystoglycane-α extracellulaire), qui s’associe avec la laminine-2, et d’une sous-unité β (dystroglycane-β) transmembranaire, qui est capable de former des complexes avec deux protéines cytoplasmiques de la cellule de Schwann, DRP2 (dystrophyn-related protein 2) et la L-périaxine [16]. Les composants de ces complexes paraissent essentiels pour le maintien de l’intégrité de la gaine de myéline.
Les souris dont le gène codant pour la L-périaxine a été invalidé développent en effet une gaine de myéline normale, mais instable, ce phénomène conduisant à l’apparition progressive d’une neuropathie démyélinisante [17]. De leur côté, des mutations récessives du gène prx, codant pour la périaxine, ont été associées chez l’homme à deux types de neuropathies démyélinisantes héréditaires, une forme 4F de CMT et le syndrome récessif de Dejerine-Sottas (DSS) [18, 19]. Par ailleurs, les souris dont le gène codant pour le dystroglycane dans les cellules de Schwann a été invalidé montrent une désorganisation importante des microvillosités au niveau des noeuds de Ranvier, une diminution de la densité de canaux Na+ et un repliement de la myéline [20]. Le dystroglycane-α semble également être le récepteur de Mycobacterium leprae, un pathogène intracellulaire des cellules de Schwann qui induit une neuropathie périphérique [21]. Enfin, des mutations du gène LAMA2, codant pour la chaîne α2 de la laminine-2, sont responsables de formes classiques de dystrophie musculaire congénitale (CMD) associées à une démyélinisation des nerfs périphériques moteurs chez l’homme et chez la souris dy [22].
Interactions axo-gliales
Au cours du développement, l’enroulement de la cellule de Schwann autour de l’axone conduit à la formation de trois types de contacts entre la cellule de Schwann et l’axone, qui définissent trois domaines anatomiquement et fonctionnellement distincts le long de l’axone : le noeud, le paranoeud et le juxtaparanoeud (Figure 2). Le noeud, caractérisé par sa forte concentration en canaux Na+, est recouvert par les microvillosités des cellules de Schwann dans le SNP. Au niveau des paranoeuds, l’axolemme est étroitement associé aux boucles latérales des cellules gliales myélinisantes, enroulées autour de l’axone, par des jonctions de type « septé » qui ressemblent aux jonctions septées des invertébrés. Au niveau de ces jonctions, l’espace intercellulaire est étroit et régulièrement interrompu par des structures denses aux électrons (bandes transverses). Ces structures semblent fortement associées au cytosquelette glial et axonal. Le juxtaparanoeud constitue une région intermédiaire entre le paranoeud et l’internoeud, où les membranes gliale et axonale sont moins étroitement associées. Ce domaine est caractérisé par son enrichissement en canaux K+ de type Shaker (sous-unités Kv1.1, Kv1.2).
Les échafaudages moléculaires intercellulaires identifiés dans les paranoeuds et les juxtaparanoeuds mettent en évidence une grande conservation des protéines constituantes et de leur organisation (Figure 4A et B) (pour revue, voir [23]). Ils sont composés d’une protéine axonale transmembranaire de la famille neurexine IV-caspr (contactin-associated protein)-paranodine (NCP), associée en cis et en trans avec des molécules d’adhérence cellulaire de la superfamille des immunoglobulines (IgSF-CAM), et sont connectés au cytosquelette axonal par l’intermédiaire de protéines de la superfamille 4.1 (4.1B). Dans les juxtaparanoeuds, le complexe intercellulaire est en outre associé aux canaux K+, probablement par l’intermédiaire d’une protéine cytoplasmique encore non identifiée.
Figure 4
Organisation moléculaire des interactions axo-gliales dans le système nerveux périphérique.
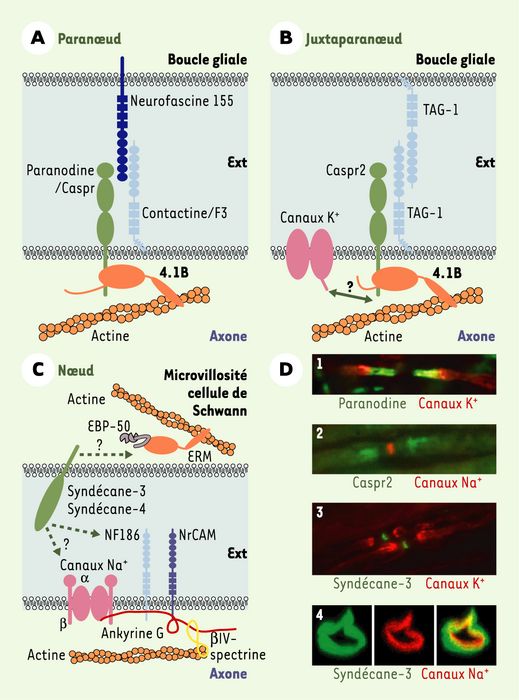
A. Complexe paranodal. Le complexe intercellulaire majeur identifié dans les paranoeuds est composé d’une glycoprotéine transmembranaire de la famille NCP (neurexine IV-caspr [contactin-associated protein]-paranodine), la paranodine/Caspr, associée en cis et en trans à deux protéines de la superfamille des immunoglobulines (IgSF-CAM), la contactine/F3 et l’isoforme de 155 kDa de la neurofascine, respectivement. Ce complexe est associé au cytosquelette d’actine axonal par l’intermédiaire de la protéine cytoplasmique 4.1B. B. Complexe juxtaparanodal. Le complexe intercellulaire majeur identifié au niveau des juxtaparanoeuds présente une composition similaire à celui identifié dans les paranoeuds : une glycoprotéine transmembranaire de la famille NCP, Caspr2, s’associe en cis et en trans avec une protéine IgSF-CAM, TAG-1 (transiently expressed axonal glycoprotein 1), qui est exprimée à la fois par la cellule gliale et le neurone ; le complexe est connecté au cytosquelette d’actine axonal par l’intermédiaire de la protéine 4.1B. Le complexe serait également associé aux canaux K+ de type Shaker par l’intermédiaire d’une protéine intracellulaire encore non identifiée. C. Protéines enrichies au niveau du noeud. Dans la membrane axonale nodale, sont enrichis les canaux Na+ dépendants du voltage et des protéines de la superfamille des immunoglobulines (IgSF-CAM), NF186 (isoforme de 186 kDa de la neurofascine) et NrCAM (NgCAM-related cell adhesion molecule), connectées au cytosquelette d’actine par l’intermédiaire de l’ankyrine G et de la βIV-spectrine. Dans les microvillosités des cellules de Schwann, sont concentrées les protéines ERM (ezrine, radixine, moésine) qui sont capables de s’associer avec l’actine et deux protéoglycanes héparane sulfate transmembranaires, le syndécane-3 et le syndécane-4. Ces derniers pourraient être impliqués dans les interactions avec les protéines enrichies de l’axone et, de plus, être connectés au cytosquelette d’actine glial en s’associant par leur région intracellulaire avec les protéines ERM. D. Exemples de localisation par immunfluorescence de protéines au niveau des jonctions axo-gliales des noeuds de Ranvier. Des anticorps capables de se fixer spécifiquement sur les protéines d’intérêt ont été appliqués sur des coupes (10 μm) de nerfs sciatiques de rat, puis leur fixation a été révélée par l’application d’un deuxième anticorps couplé à une molécule fluorescente. 1. Immunolocalisation de paranodine/Caspr dans les paranoeuds (vert) et des canaux K+ dans les juxtaparanoeuds (rouge). 2. Immunolocalisation de Caspr2 dans les juxtaparanoeuds (vert) et des canaux Na+ dans le noeud (rouge). 3. Immunolocalisation du syndécane-3 dans les microvillosités des cellules de Schwann (vert) et des canaux K+ dans les juxtaparanoeuds (rouge). 4. Immunolocalisation du syndécane-3 dans les microvillosités des cellules de Schwann (vert) et des canaux Na+ dans l’axone au niveau du noeud (rouge) (1-3 : coupes longitudinales d’une fibre nerveuse ; 4 : coupe transversale d’une fibre nerveuse au niveau du noeud). Ext : espace intercellulaire ; EBP-50 : ERM-binding phosphoprotein 50 kDa.
Au niveau des noeuds, les canaux Na+ font également partie de complexes axonaux multimoléculaires connectés au cytosquelette, comprenant plusieurs protéines extracellulaires, transmembranaires et intracellulaires (Figure 4C) (pour revue, voir [23]). Les principales molécules transmembranaires sont des protéines IgSF-CAM. Ces molécules et les sous-unités β des canaux Na+ s’associent avec l’ankyrine G, elle-même associée à la βIV-spectrine, créant ainsi un lien avec le cytosquelette d’actine. L’enrichissement de toutes ces protéines au niveau du noeud requiert dans le SNP un contact direct entre l’axone et la cellule de Schwann. Cependant, les molécules gliales impliquées dans ce processus ne sont pas connues. Deux protéoglycanes héparane sulfate transmembranaires, le syndécane-3 et le syndécane-4, sont enrichis dans les microvillosités des cellules de Schwann [24]. Ce sont des molécules qui ont été impliquées dans l’adhérence entre les cellules ou entre les cellules et la matrice extracellulaire.
L’étude de modèles animaux murins, dont les gènes codant pour la paranodine/Caspr ou la contactine/F3 - les protéines axonales des familles NCP et IgSF-CAM, respectivement, présentes au niveau des paranoeuds - ont été invalidés, montre que ces deux protéines sont essentielles pour la formation des jonctions de type « septé » [25, 26]. Une perturbation de ces jonctions a, par ailleurs, été observée chez d’autres souris mutantes [27-29]. Dans tous les cas, la disparition des jonctions paranodales résulte en une perturbation générale de l’organisation moléculaire des fibres myélinisées au niveau des noeuds de Ranvier, dont une délocalisation des canaux K+ des juxtaparanoeuds vers les paranoeuds, la myéline compacte restant par ailleurs intacte. Ces altérations structurales paranodales se traduisent, sur le plan physiologique, par une diminution de la vitesse de conduction de l’influx nerveux. Ces observations suggèrent que les jonctions paranodales sont essentielles pour la structure et la fonction des fibres myélinisées en jouant un rôle de séparation des protéines membranaires axonales du noeud de celles de l’internoeud. Il a été proposé que ces jonctions puissent également assurer une fonction de barrière, ralentissant la diffusion des molécules dans l’espace intercellulaire de la région nodale vers l’internoeud. L’étude de modèles animaux murins dont les gènes codant pour les protéines NCP et IgSF-CAM juxtaparanodales ont été invalidés - Caspr2 et TAG-1 (transiently expressed axonal glycoprotein 1), respectivement - a par ailleurs montré que ces protéines sont essentielles pour une localisation et une concentration correctes des canaux K+ dans les juxtaparanoeuds [30, 31]. Cette observation est intéressante sur le plan physiologique, dans la mesure où il a été proposé que les canaux K+ juxtaparanodaux puissent participer au rétablissement rapide du potentiel de repos.
Conclusions
Dans le système nerveux périphérique, les cellules de Schwann myélinisantes et les neurones échangent des signaux de survie et de différenciation tout au long du développement. Des signaux émis par l’axone dirigent la différenciation et la réorganisation de la cellule de Schwann qui, en retour, remodèle l’axone en une série de domaines polarisés différant dans leur structure, leur fonction et leur organisation moléculaire. Une telle situation laisse prédire que des altérations dans les cellules de Schwann myélinisantes vont presque invariablement affecter leur partenaire neuronal, et inversement.
L’observation de nombreux cas cliniques de neuropathies périphériques démyélinisantes renforce cette hypothèse : des altérations de la cellule de Schwann induisent une démyélinisation associée à une modification de la vitesse de conduction des nerfs, qui, finalement, conduit à une perte axonale. Dans ce contexte, il apparaît essentiel de comprendre les mécanismes normaux contrôlant les interactions des cellules gliales myélinisantes entre elles, avec les neurones, et avec la matrice extracellulaire. Des composants majeurs de ces interactions ont été identifiés ces dernières années. Certains des composants gliaux ont été impliqués dans des neuropathies périphériques démyélinisantes. Les conséquences sur l’axone et les interactions axo-gliales n’ont cependant pas été étudiées sur un plan moléculaire. Aucun composant neuronal impliqué dans les interactions axo-gliales n’a par ailleurs encore été associé à une neuropathie particulière. Des recherches dans ces différents domaines devraient permettre de mieux comprendre la physiopathologie des multiples neuropathies périphériques et, à terme, d’envisager des approches thérapeutiques efficaces.
Parties annexes
Références
- 1. Arroyo EJ, Scherer SS. On the molecular architecture of myelinated fibers. Histochem Cell Biol 2000 ; 113 : 1-18.
- 2. Filbin MT, Tennekoon GI. The role of complex carbohydrates in adhesion of the myelin protein, P0. Neuron 1991 ; 7 : 845-55.
- 3. Tetzlaff W. Tight junction contact events and temporary gap junctions in the sciatic nerve fibres of the chicken during Wallerian degeneration and subsequent regeneration. J Neurocytol 1982 ; 11 : 839-58.
- 4. Fannon AM, Sherman DL, Ilyina-Gragerova G, et al. Novel E-cadherin-mediated adhesion in peripheral nerve : Schwann cell architecture is stabilized by autotypic adherens junctions. J Cell Biol 1995 ; 129 : 189-202.
- 5. Gow A, Southwood CM, Li JS, et al. CNS myelin and Sertoli cell tight junction strands are absent in Osp/claudin-11 null mice. Cell 1999 ; 99 : 649-59.
- 6. Tsukita S, Furuse M. Claudin-based barrier in simple and stratified cellular sheets. Curr Opin Cell Biol 2002 ; 14 : 531-6.
- 7. Poliak S, Matlis S, Ullmer C, et al. Distinct claudins and associated PDZ proteins form different autotypic tight junctions in myelinating Schwann cells. J Cell Biol 2002 ; 159 : 361-72.
- 8. Nicholson BJ. Gap junctions : from cell to molecule. J Cell Sci 2003 ; 116 : 4479-81.
- 9. Balice-Gordon RJ, Bone LJ, Scherer SS. Functional gap junctions in the schwann cell myelin sheath. J Cell Biol 1998 ; 142 : 1095-104.
- 10. Altevogt BM, Kleopa KA, Postma FR, et al. Connexin29 is uniquely distributed within myelinating glial cells of the central and peripheral nervous systems. J Neurosci 2002 ; 22 : 6458-70.
- 11. Scherer SS, Deschenes SM, Xu YT, et al. Connexin32 is a myelin-related protein in the PNS and CNS. J Neurosci 1995 ; 15 : 8281-94.
- 12. Suter U, Scherer SS. Disease mechanisms in inherited neuropathies. Nat Rev Neurosci 2003 ; 4 : 714-26.
- 13. Berger P, Young P, Suter U. Molecular cell biology of Charcot-Marie-Tooth disease. Neurogenetics 2002 ; 4 : 1-15.
- 14. Young P, Boussadia O, Berger P, et al. E-cadherin is required for the correct formation of autotypic adherens junctions of the outer mesaxon but not for the integrity of myelinated fibers of peripheral nerves. Mol Cell Neurosci 2002 ; 21 : 341-51.
- 15. Yamada H, Denzer AJ, Hori H, et al. Dystroglycan is a dual receptor for agrin and laminin-2 in Schwann cell membrane. J Biol Chem 1996 ; 271 : 23418-23.
- 16. Sherman DL, Fabrizi C, Gillespie CS, et al. Specific disruption of a Schwann cell dystrophin-related protein complex in a demyelinating neuropathy. Neuron 2001 ; 30 : 677-87.
- 17. Gillespie CS, Sherman DL, Fleetwood-Walker SM, et al. Peripheral demyelination and neuropathic pain behavior in periaxin-deficient mice. Neuron 2000 ; 26 : 523-31.
- 18. Boerkoel CF, Takashima H, Stankiewicz P, et al. Periaxin mutations cause recessive Dejerine-Sottas neuropathy. Am J Hum Genet 2001 ; 68 : 325-33.
- 19. Guilbot A, Williams A, Ravise N, et al. A mutation in periaxin is responsible for CMT4F, an autosomal recessive form of Charcot-Marie-Tooth disease. Hum Mol Genet 2001 ; 10 : 415-21.
- 20. Saito F, Moore SA, Barresi R, et al. Unique role of dystroglycan in peripheral nerve myelination, nodal structure, and sodium channel stabilization. Neuron 2003 ; 38 : 747-58.
- 21. Rambukkana A. Mycobacterium leprae-induced demyelination : a model for early degeneration. Curr Opin Immunol 2004 ; 16 : 511-8.
- 22. Matsumura K, Yamada H, Saito F, et al. Peripheral nerve involvement in merosin-deficient cognenital muscular dystrophy and dy mouse. Neuromusc. Disorders 1997 ; 7 :7-12.
- 23. Salzer JL. Polarized domains of myelinated axons. Neuron 2003 ; 40 : 297-318.
- 24. Goutebroze L, Carnaud M, Denisenko N, et al. Syndecan-3 and syndecan-4 are enriched in Schwann cell perinodal processes. BMC Neurosci 2003 ; 4 : 29.
- 25. Bhat MA, Rios JC, Lu Y, et al. Axon-glia interactions and the domain organization of myelinated axons require neurexin IV/Caspr/Paranodin. Neuron 2001 ; 30 : 369-83.
- 26. Boyle ME, Berglund EO, Murai KK, et al. Contactin orchestrates assembly of the septate-like junctions at the paranode in myelinated peripheral nerve. Neuron 2001 ; 30 : 385-97.
- 27. Dupree JL, Girault JA, Popko B. Axo-glial interactions regulate the localization of axonal paranodal proteins. J Cell Biol 1999 ; 147 : 1145-52.
- 28. Ishibashi T, Dupree JL, Ikenaka K, et al. A myelin galactolipid, sulfatide, is essential for maintenance of ion channels on myelinated axon but not essential for initial cluster formation. J Neurosci 2002 ; 22 : 6507-14.
- 29. Poliak S, Gollan L, Salomon D, et al. Localization of Caspr2 in myelinated nerves depends on axon-glia interactions and the generation of barriers along the axon. J Neurosci 2001 ; 21 : 7568-75.
- 30. Poliak S, Salomon D, Elhanany H, et al. Juxtaparanodal clustering of Shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J Cell Biol 2003 ; 162 : 1149-60.
- 31. Traka M, Goutebroze L, Denisenko N, et al. Association of TAG-1 with Caspr2 is essential for the molecular organization of juxtaparanodal regions of myelinated fibers. J Cell Biol 2003 ; 162 : 1161-72.
Liste des figures
Figure 1
Conduction de l’influx nerveux dans les fibres myélinisées et non myélinisées.
Les potentiels d’action transmis le long des axones sont produits par un flux d’ions à travers des canaux Na+ dépendants du voltage, qui modifie la distribution des charges de part et d’autre de la membrane. A. Dans les fibres non myélinisées, les canaux Na+ sont répartis de façon uniforme dans la membrane axonale, et le potentiel d’action est transmis de façon continue le long de l’axone à une vitesse ne dépassant pas 3 m/s. B. Dans les fibres myélinisées, les canaux Na+ sont concentrés dans des régions discontinues de l’axone, appelées noeuds de Ranvier, permettant une propagation rapide, dite « saltatoire », du potentiel d’action par bonds d’un noeud à l’autre. Les noeuds de Ranvier sont séparés par une gaine de myéline isolante, formée par les cellules de Schwann dans le système nerveux périphérique, et par les oligodendrocytes dans le système nerveux central.
Figure 2
Organisation ultrastructurale d’une fibre myélinisée du système nerveux périphérique.
La myéline se divise en deux sous-domaines structurellement et fonctionnement distincts : la myéline compacte et la myéline non compacte. La myéline compacte est localisée dans les internoeuds (IN) situés entre les noeuds de Ranvier, et résulte de l’accolement des membranes adjacentes de la cellule de Schwann lors de son enroulement progressif autour de l’axone. Dans ces régions, la myéline ne contient plus de cytoplasme. La myéline non compacte correspond, quant à elle, à des régions de la cellule de Schwann où persiste du cytoplasme. Ces régions correspondent aux boucles gliales paranodales, aux incisures de Schmidt-Lanterman, aux régions péri-axonale et ab-axonale et aux microvillosités. Au cours du développement, l’enroulement de la cellule de Schwann autour de l’axone conduit à la formation de trois types de contacts entre la cellule de Schwann et l’axone, qui définissent trois domaines anatomiquement et fonctionnellement distincts le long de l’axone : le noeud (N), le paranoeud (PN) et le juxtaparanoeud (JPN). Le noeud est recouvert par les microvillosités des cellules de Schwann. Au niveau des paranoeuds, l’axolemme est étroitement associé aux boucles latérales des cellules gliales myélinisantes par des jonctions de type « septé ». Le juxtaparanoeud correspond à une région intermédiaire entre le paranoeud et l’internoeud. Partie supérieure de la figure : photo en microscopie électronique d’un noeud de Ranvier d’une fibre myélinisée de nerf phrénique (échelle, 2 μm).
Figure 3
Organisation moléculaire des interactions gliales autotypiques et de la connexion ente la cellule gliale et la lame basale, dans le système nerveux périphérique.
A. Protéines majeures de la myéline compacte. Les trois protéines majeures de la myéline compacte sont les protéines P0 (myelin protein zero), PMP22 (peripheral myelin protein 22) et MBP (myelin basic protein). La protéine P0 joue le rôle de molécule d’adhérence grâce à la capacité de son domaine extracellulaire de type « immunoglobuline » d’établir des interactions homotypiques en cis et en trans. B. Protéines impliquées dans les jonctions autotypiques de la myéline non compacte. Dans les boucles paranodales, les incisures de Schmidt-Lanterman et les régions péri-axonale et ab-axonale, les membranes plasmiques des cellules de Schwann myélinisantes établissent des jonctions autotypiques dites « adhérentes » et « serrées ». Dans les boucles paranodales et les incisures se situent également des jonctions dites « communicantes ». Les jonctions adhérentes contiennent une molécule d’adhérence dépendante du calcium, la E-cadhérine, ainsi que la β-caténine cytoplasmique qui connecte la E-cadhérine aux filaments d’actine. Les protéines identifiées dans les jonctions serrées sont des protéines membranaires intégrales, la claudine-1 et la claudine-5. Les jonctions communicantes se forment par la fusion de deux hémicanaux correspondant à un assemblage de 6 connexines identiques, la connexine 32 (Cx32) ou la connexine 29 (Cx29). C. Protéines impliquées dans la connexion entre la cellule de Schwann myélinisante et sa lame basale. La laminine-2 constitue l’un des composants majeurs de la lame basale. L’un de ses récepteurs sur la cellule de Schwann est le dystroglycane, capable de former des complexes avec deux protéines cytoplasmiques de la cellule de Schwann, DRP2 (dystrophin-related protein 2) et la L-périaxine. Int et Ext : intérieur et extérieur de la cellule de Schwann.
Figure 4
Organisation moléculaire des interactions axo-gliales dans le système nerveux périphérique.
A. Complexe paranodal. Le complexe intercellulaire majeur identifié dans les paranoeuds est composé d’une glycoprotéine transmembranaire de la famille NCP (neurexine IV-caspr [contactin-associated protein]-paranodine), la paranodine/Caspr, associée en cis et en trans à deux protéines de la superfamille des immunoglobulines (IgSF-CAM), la contactine/F3 et l’isoforme de 155 kDa de la neurofascine, respectivement. Ce complexe est associé au cytosquelette d’actine axonal par l’intermédiaire de la protéine cytoplasmique 4.1B. B. Complexe juxtaparanodal. Le complexe intercellulaire majeur identifié au niveau des juxtaparanoeuds présente une composition similaire à celui identifié dans les paranoeuds : une glycoprotéine transmembranaire de la famille NCP, Caspr2, s’associe en cis et en trans avec une protéine IgSF-CAM, TAG-1 (transiently expressed axonal glycoprotein 1), qui est exprimée à la fois par la cellule gliale et le neurone ; le complexe est connecté au cytosquelette d’actine axonal par l’intermédiaire de la protéine 4.1B. Le complexe serait également associé aux canaux K+ de type Shaker par l’intermédiaire d’une protéine intracellulaire encore non identifiée. C. Protéines enrichies au niveau du noeud. Dans la membrane axonale nodale, sont enrichis les canaux Na+ dépendants du voltage et des protéines de la superfamille des immunoglobulines (IgSF-CAM), NF186 (isoforme de 186 kDa de la neurofascine) et NrCAM (NgCAM-related cell adhesion molecule), connectées au cytosquelette d’actine par l’intermédiaire de l’ankyrine G et de la βIV-spectrine. Dans les microvillosités des cellules de Schwann, sont concentrées les protéines ERM (ezrine, radixine, moésine) qui sont capables de s’associer avec l’actine et deux protéoglycanes héparane sulfate transmembranaires, le syndécane-3 et le syndécane-4. Ces derniers pourraient être impliqués dans les interactions avec les protéines enrichies de l’axone et, de plus, être connectés au cytosquelette d’actine glial en s’associant par leur région intracellulaire avec les protéines ERM. D. Exemples de localisation par immunfluorescence de protéines au niveau des jonctions axo-gliales des noeuds de Ranvier. Des anticorps capables de se fixer spécifiquement sur les protéines d’intérêt ont été appliqués sur des coupes (10 μm) de nerfs sciatiques de rat, puis leur fixation a été révélée par l’application d’un deuxième anticorps couplé à une molécule fluorescente. 1. Immunolocalisation de paranodine/Caspr dans les paranoeuds (vert) et des canaux K+ dans les juxtaparanoeuds (rouge). 2. Immunolocalisation de Caspr2 dans les juxtaparanoeuds (vert) et des canaux Na+ dans le noeud (rouge). 3. Immunolocalisation du syndécane-3 dans les microvillosités des cellules de Schwann (vert) et des canaux K+ dans les juxtaparanoeuds (rouge). 4. Immunolocalisation du syndécane-3 dans les microvillosités des cellules de Schwann (vert) et des canaux Na+ dans l’axone au niveau du noeud (rouge) (1-3 : coupes longitudinales d’une fibre nerveuse ; 4 : coupe transversale d’une fibre nerveuse au niveau du noeud). Ext : espace intercellulaire ; EBP-50 : ERM-binding phosphoprotein 50 kDa.
Liste des tableaux
Tableau I
Importance fonctionnelle des contacts cellulaires dans les fibres myélinisées du système nerveux périphérique.
CMT : maladie de Charcot-Marie-Tooth ; DSS : neuropathie récessive de Dejerine-Sottas ; CH : hypomyélinisation congénitale ; CMD : dystrophie musculaire congénitale ; KO : souris dont les gènes ont été invalidés ; Tg : souris transgéniques ; Mut : souris présentant des mutations spontanées ; PMP22 : peripheral myelin protein 22 ; PO : myelin protein zero ; Cx : connexine ; Caspr : contactin-associated protein ; TAG-1 : transiently-expressed axonal glycoprotein 1.