Résumés
Résumé
Le diagnostic présymptomatique en neurogénétique concerne un nombre croissant d’affections, principalement neurodégénératives, au premier rang desquelles la maladie de Huntington. La possibilité pour une personne à risque de connaître son statut génétique vis-à-vis d’une maladie connue dans la famille pose des problèmes puisque, dans la plupart des cas, il n’existe ni prévention, ni traitement possibles en cas de résultat défavorable. De plus, le fait d’être porteur ne dit pas à quel moment et sous quelle forme la maladie se manifestera, d’où une limitation importante pour planifier l’avenir. Seule une minorité des personnes à risque choisissent de faire le test. Notre expérience souligne l’intérêt d’une prise en charge pluridisciplinaire au long cours des candidats à un test présymptomatique : si celle-ci est indispensable pour le choix éclairé et autonome du candidat au test, elle n’empêche toutefois pas des changements importants, parfois radicaux, en cas de résultat défavorable, bien sûr, mais également après un résultat établissant un statut de non-porteur. Enfin, l’expérience acquise pour la maladie de Huntington permet maintenant d’aborder le test présymptomatique dans d’autres affections ; malgré des différences dans la présentation et l’évolution de ces maladies, les conséquences du test en l’absence de thérapeutique spécifique restent similaires.
Summary
Presymptomatic testing is available since 15 years for Huntington disease and it is now possible for a number of other neurogenetic disorders, mostly neurodegenerative disorders. The possibility of determining the genetic status of an at-risk person for the disorder which run in his family raises questions because of the absence of preventive and curative treatments in most instances. In addition, being carrier does not tell you when the disease will start and how it will evolve, impairing the possibilities of planning the future. A pluridisciplinary approach to predictive testing with care before, during and after the test taking into account the medical, social and psychological aspects of the disease is good practice. At the present time, only a minority of at-risk individuals request presymptomatic testing and almost 50 % do not pursue until the results. The consequences of the test may be harmful, more frequently after an unfavorable than after a favorable result. Although the motivations and the outcome in terms of request for prenatal testing after a carrier result are different in Huntington’s disease and spinocerebellar ataxias, our experience underlines the benefit of pluridisciplinary care and of time for decision taking. For other disorders like familial Alzheimer’s disease, or familial Creutzfeldt-Jakob disease, the experience in presymptomatic testing is still limited but the situation seems similar to Huntington’s disease because of the presence of dementia. It will be interesting to study the motivations and the outcome of the tests in disorders like autosomal dominant spastic paraplegias which usually do not reduce the life expectancy. Nevertheless, the overall situation might change greatly when efficient treatments will become available in these disorders.
Corps de l’article
Un des faits nouveaux en génétique médicale est d’avoir montré que les maladies héréditaires à révélation tardive sont plus nombreuses et plus fréquentes que l’on croyait. On peut estimer qu’environ 1 %, peut-être plus, de la population d’âge adulte est concerné par ces pathologies, pour la plupart de nature neurodégénérative. De nombreux gènes ont déjà été identifiés, et l’hétérogénéité génétique s’est révélée très importante. Ces connaissances génétiques permettent un diagnostic présymptomatique, par analyse moléculaire, de ces affections chez le sujet à risque élevé. Certaines de ces maladies sont monogéniques dans la totalité, ou la quasi totalité, des cas (maladie de Huntington, dystrophie myotonique de Steinert), tandis que d’autres sont des sous-entités mendéliennes de maladies qui, dans la grande majorité des cas, seront multifactorielles (démences de type Alzheimer, maladie de Parkinson) (Tableau I). Il est cependant troublant de noter que les formes héréditaires monogéniques de la maladie de Parkinson ne peuvent être distinguées des formes dites sporadiques, ou idiopathiques, aussi bien pour ce qui concerne l’âge de début de la maladie que sa progression. Ces maladies se transmettent dans la majorité des cas selon un mode dominant autosomique, parfois avec une pénétrance réduite. Certaines sont très graves, et ne bénéficient d’aucun traitement préventif ou curatif, d’autres peuvent être traitées, ou prévenues, avec une efficacité variable selon la maladie.
Tableau I
Exemples de maladies neurodégénératives pour lesquelles il existe des formes héréditaires et où un test prédictif est possible dans certains sous-groupes génétiques.

De façon schématique, un test génétique peut être proposé dans trois situations différentes. La première vise à confirmer le diagnostic d’une maladie génétique chez une personne ayant des symptômes ; le test peut alors être considéré comme un moyen de porter avec certitude un diagnostic évoqué cliniquement. Dans un deuxième cas, il s’agit de « faire un test génétique » avant que des signes de la maladie héréditaire n’apparaissent : les maladies neurologiques à révélation tardive peuvent susciter ce type de demande de diagnostic présymptomatique ; être porteur, développer la maladie et risquer de la transmettre, ou ne pas être porteur et donc ne pas transmettre la maladie sont les deux résultats possibles. Enfin, la question d’un diagnostic prénatal peut être posée pour un foetus à risque, même s’il s’agit d’un acte controversé et difficile dans les maladies qui ne se manifestent qu’à l’âge adulte. Cette revue se concentrera sur les tests présymptomatiques et leurs conséquences sur les demandes de diagnostic prénatal.
Les interrogations préalables au test
Plusieurs niveaux de discussion se proposent aux personnes à risque, non malades, qui ont le souhait de faire un test présymptomatique.
Quelles seront les possibilités de traitement après un test présymptomatique ?
La question de « faire, ou ne pas faire, un test prédictif » s’organise autour des possibilités de traitement ou de prévention. Il serait idéal de transformer un test présymptomatique en test de « dépistage », un terme qui implique la mise en place d’un traitement ou d’une prévention comme conséquence immédiate du test : la question de faire ou de ne pas faire serait alors obsolète. Les possibilités de prévention vont du dépistage, parfois associé à une prévention efficace (chirurgie préventive des cancers de la thyroïde dans la néoplasie endocrinienne multiple de type II, par exemple), jusqu’au test « brut », sans possibilité de traitement et de prévention après un résultat défavorable, notamment dans le cas des maladies neurodégénératives (dont l’exemple le plus connu est celui de la maladie de Huntington, mais d’autres exemples sont présentés dans le Tableau I). Entre ces deux extrêmes, les maladies s’échelonnent selon les possibilités de prévention ou de traitement plus au moins efficaces (surveillance renforcée, voire mastectomie et ovariectomie chez les femmes porteuses d’une mutation de BRCA1, notamment) : le degré d’intervention varie donc selon la pathologie (Figure 1).
Figure 1
Bénéfice médical du diagnostic présymptomatique des maladies héréditaires.
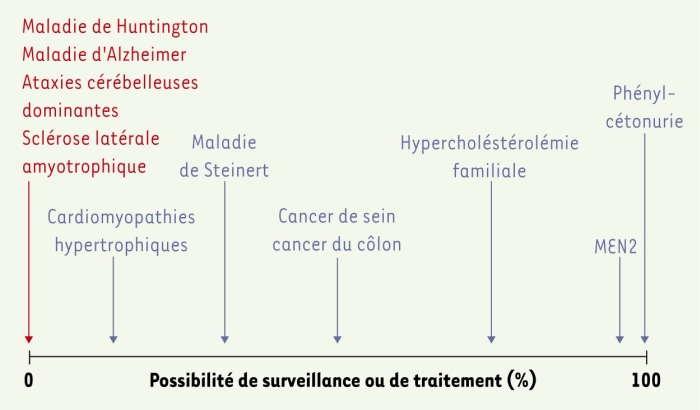
Le bénéfice médical d’un diagnostic présymptomatique varie selon que l’on dispose ou non de méthodes de surveillance, de prévention ou de traitement pour la maladie (voir texte). MEN2 : néoplasies endocrines multiples de type 2.
La prise de décision chez la personne à risque prendra en compte les possibilités de prévention et le vécu de la maladie chez le membre atteint de la famille : une maladie jugée relativement bénigne pour les médecins, mais associée à un vécu douloureux individuel et familial, peut en effet être à l’origine de demandes d’un test présymptomatique, voire prénatal.
Quel est le pouvoir prédictif du test présymptomatique, et quelles en sont les limites ?
Selon la maladie, le pouvoir prédictif du test est très différent. A priori, la valeur prédictive est claire pour les maladies monogéniques ayant une pénétrance complète : un sujet porteur de la mutation développera la maladie. Mais quand, et comment ? L’âge de début de la maladie est souvent très variable pour les maladies à transmission dominante, même à révélation tardive : en moyenne entre 30 et 50 ans, mais des formes apparaissant après 60 ans ne sont pas rares. Par ailleurs, une pénétrance incomplète de la mutation et une expressivité clinique variable de la maladie affaiblit le pouvoir prédictif des tests pour beaucoup de maladies dominantes à révélation tardive. Le test génétique ne permet donc pas réellement de prédire précisément l’avenir de la personne porteuse : quels seront l’âge de début, l’évolution et la gravité de la maladie ? Le test génétique détermine un génotype, et non un phénotype. Ainsi, le pouvoir prédictif du test génétique est à la fois fort, donnant une quasi certitude d’être atteint en cas de résultat défavorable, mais aussi faible, car incapable de prédire le début et l’évolution de la maladie.
Dans les maladies multifactorielles, qui relèvent de facteurs génétiques et de facteurs environnementaux, le pouvoir prédictif du test génétique est très faible. En effet, les facteurs génétiques impliqués sont largement répandus dans la population générale, et bon nombre de personnes porteuses d’un de ces facteurs ne développeront jamais la maladie. Dans le cas de la maladie d’Alzheimer, par exemple, l’allèle E4 de l’apolipoprotéine (ApoE4) est un facteur de risque important, puisqu’il augmente le risque de maladie d’un facteur 2 à 3 chez les sujets hétérozygotes, et 7 à 9 chez les homozygotes. Mais le test génétique est peu contributif pour confirmer la maladie chez une personne malade, puisque d’authentiques patients ayant une maladie d’Alzheimer ne portent pas l’allèle E4. Pour l’instant, le test génétique et la révélation de ce facteur de risque n’ont aucune utilité clinique chez une personne saine. La prédiction est donc très relative : il s’agit plus de la révélation d’un statut génétique que d’un état clinique, et les personnes à risque doivent connaître les incertitudes médicales quant à la prédiction per se.
Le diagnostic présymptomatique ne se conçoit donc actuellement que chez une personne ayant un risque accru vis-à-vis d’une affection monogénique, qui souhaite réellement connaître son statut génétique après que l’affection ait été identifiée chez un apparenté, en sachant qu’elle n’en tirera pas de bénéfice médical immédiat et qu’elle doit accepter les incertitudes cliniques de la prédiction.
La maladie de Huntington : un paradigme dans la pratique des tests présymptomatiques
La maladie de Huntington a donné lieu à la réflexion la plus approfondie et bénéficie du plus grand recul dans la pratique de tests présymptomatiques. Cette maladie est la première pour laquelle un test présymptomatique devint techniquement possible, d’abord par analyse indirecte, dès la fin des années 80, puis par analyse directe, fiable, depuis 1993 et la découverte du gène responsable et de sa mutation causale.
La maladie de Huntington est une maladie à transmission autosomique dominante, dans laquelle une histoire familiale de troubles psychiatriques, cognitifs et comportementaux et/ou de mouvements anormaux est très souvent retrouvée. Il s’agit d’une maladie héréditaire rare, dont la prévalence est estimée à 0,5 à 1/10 000 en Europe, mais qui touche toutes les ethnies [1]. La maladie de Huntington est liée à l’expansion anormale d’un trinucléotide CAG dans le premier exon du gène IT15, situé sur le bras court du chromosome 4, qui code pour la huntingtine. Le nombre de répétitions est inférieur à 30 sur les chromosomes normaux, et supérieur ou égal à 36 chez les patients. Dans la majorité des cas, la longueur de l’expansion se situe entre 40 et 45 CAG. Il existe une corrélation inverse entre nombre de CAG et l’âge de début, la sévérité clinique et l’intensité des lésions neuropathologiques, mais avec d’importantes variations individuelles. Si l’âge de début diminue avec le nombre de répétitions, ce nombre ne permet toutefois pas de prédire précisément l’âge de début ou la rapidité d’aggravation d’un patient. Dans la plupart des cas, l’instabilité de la répétition pendant la transmission a pour conséquence l’augmentation, de quelques unités, du nombre des triplets CAG chez l’enfant porteur ; dans quelques rares cas, la transmission paternelle s’accompagne d’une grande augmentation de la longueur de l’expansion, qui peut alors dépasser 60 répétitions CAG et être à l’origine de cas juvéniles de la maladie.
L’hétérogénéité génétique de la maladie de Huntington a été démontrée avec la découverte d’un second gène, JPH3 [2]. Les phénotypes liés à une mutation de IT15 ou de JPH3 ne sont pas cliniquement distinguables, mais il a été montré que JPH3 est beaucoup plus fréquemment associé à une maladie de Huntington chez les patients d’origine africaine (30 % en Afrique du Sud parmi les formes cliniques de maladie de Huntington [3]). Par ailleurs, les formes adultes de l’atrophie dentato-rubropallido-luysienne (DRPLA), beaucoup plus fréquentes au Japon qu’en Europe, peuvent mimer une maladie de Huntington.
Règles de bonnes pratiques
Une réflexion internationale a conduit à l’élaboration de règles qui encadrent les bonnes pratiques du diagnostic présymptomatique de la maladie de Huntington [4]. Cinq principes sont mis en avant : bénéfice, autonomie, choix éclairé, confidentialité et égalité. Dans ce cadre, le bénéfice n’est pas thérapeutique et dépend de la demande individuelle de la personne à risque. Le principe d’autonomie requiert que le test ne soit demandé qu’à titre individuel, et par une personne majeure. Le choix éclairé nécessite de délivrer une information aussi complète que possible sur la maladie et ses caractéristiques génétiques, ainsi que les différentes options en matière de test. La confidentialité est capitale pour l’avenir de la personne à risque, surtout si elle reçoit une réponse défavorable. Enfin, le principe d’égalité s’applique aux possibilités d’accès de la personne à risque aux centres qui pratiquent le test présymptomatique, sans discrimination de nature financière. Il est clair qu’un diagnostic présymptomatique n’est utile que s’il est à l’origine d’une prise en charge médicale et psychologique.
Composition de l’équipe et déroulement du test
Afin de prendre en compte les exigences du choix informé, le déroulement du test présymptomatique dans le temps et la prise en charge de la personne à risque par une équipe pluridisciplinaire sont particulièrement importants. La composition de l’équipe reflète les problèmes soulevés par la maladie concernée : généticien et neurologue connaissant la maladie, psychologue avertie de la transmission autosomique dominante et du devenir des personnes à risque après le test, assistante sociale au courant des enjeux sociaux et des conséquences dans le domaine des assurances et, enfin, infirmière de génétique consciente de la nécessité du déroulement des entretiens dans le temps et de la disponibilité de l’équipe. L’existence de plusieurs interlocuteurs autorise une réflexion variée grâce à leurs approches différentes de la maladie, afin de prendre en compte la spécificité de chaque demande. Il est intéressant de constater que le désir de savoir est le moteur le plus grand pour faire le test, et que même une grossesse en cours n’est pas plus incitative à faire un test [5].
Un cadre de trois entretiens après le premier contact et jusqu’à la prise de décision et le prélèvement sanguin est proposé : une consultation avec la psychologue, une avec l’assistante sociale et une dernière avec le généticien. Les personnes à risque connaissent le nom des intervenants, une brochure leur est remise avec le numéro de téléphone direct qui permet une organisation adaptée des rendez-vous successifs. La possibilité de rencontrer un psychiatre ou de faire un test de mémoire, pour détecter les premiers signes de la maladie, est également offerte.
Le décret du 23 juin 2000 limite la prescription des tests présymptomatiques à l’intervention d’équipes pluridisciplinaires, rassemblant les compétences médicales nécessaires. L’article 145-15-5 stipule ainsi : « Chez une personne asymptomatique, mais présentant des antécédents familiaux, la prescription d’un examen des caractéristiques génétiques ne peut avoir lieu que dans le cadre d’une consultation médicale individuelle. Cette consultation doit être effectuée par un médecin oeuvrant au sein d’une équipe pluridisciplinaire rassemblant des compétences cliniques et génétiques. Cette équipe doit se doter d’un protocole type de prise en charge et être déclarée au ministère chargé de la santé… ». Dans le même décret, la réalisation de ce test est interdite chez un mineur, sauf si ce dernier ou sa famille peuvent personnellement bénéficier de mesures préventives ou curatives immédiates.
Les enseignements de la pratique du test présymptomatique
Une consultation dédiée a été instaurée dès 1992 à l’Hôpital de la Salpêtrière. Aujourd’hui, 15 centres nationaux prennent en charge les demandes.
Depuis la mise en place de notre centre, 984 personnes à risque sont venues à cette consultation pour demander un test présymptomatique, 527 (55 %) ont continué la démarche en rencontrant l’ensemble des membres de la consultation, et 352 parmi elles ont obtenu le résultat. Trois chiffres soulignent l’importance du temps dans la réflexion de la personne à risque : seules 20 % des personnes à risque formulent une demande de test ; près d'une personne sur deux ne poursuit pas sa démarche après le premier entretien ; une personne sur dix choisit de suspendre sa démarche entre le premier entretien et la prise de sang [6]. Existe-t-il une différence entre ceux qui poursuivent et ceux qui ne souhaitent pas aller jusqu’au bout de leur demande de diagnostic présymptomatique ? Une étude réalisée dans notre centre a montré que ceux qui poursuivent ont plusieurs motivations, et sont plus orientés vers un projet parental, ou ont déjà des enfants, et souhaitent informer leurs enfants de leur risque. En revanche, ceux qui abandonnent ont une connaissance récente de leur risque et un nombre plus limité de motivations.
Le test présymptomatique est loin d’être un acte médical neutre : le résultat équivaut en effet soit à une condamnation, soit à une libération. Quel est l’impact du résultat et les conséquences d’un résultat défavorable ? Une étude multicentrique montre que la proportion d’événement indésirables graves, en rapport avec la réalisation du test présymptomatique, reste limitée, que le résultat soit ou non défavorable [7]. Si les sujets porteurs sont plus souvent déprimés (56 %) et vont moins bien que les non-porteurs, ces derniers sont également déprimés dans 31 % des cas et nécessitent un suivi. Cela souligne que les entretiens avec des intervenants différents, respectueux du choix de la personne à risque, jouent un rôle crucial dans la préparation au résultat et au suivi ultérieur. Même après un résultat favorable, le temps pour guérir du fait d’être à risque est important à prendre en considération [8] : ce temps, variable selon les individus, est très clairement plus bref pour le conjoint que pour la personne à risque, dont le statut « d’être à risque » fait partie de son identité. Des réactions paradoxales peuvent ainsi se produire chez les non-porteurs de la mutation après l’annonce du résultat : le remaniement identitaire est important, car certaines personnes à risque avaient construit toute leur vie en fonction de l’idée d’être porteurs, tandis que d’autres développent une culpabilité du survivant par rapport à d’autres membres de la fratrie.
Le diagnostic prénatal
Le diagnostic prénatal n’est pas une conséquence directe du test présymptomatique, comme cela a été montré par les équipes canadiennes et, plus récemment, par un groupe de travail Européen [9]. Le diagnostic prénatal dans les maladies récessives graves se justifie non seulement par le fait que le risque pour la descendance n’est que de 25 %, mais aussi parce que les parents ne seront pas atteints de la maladie. La situation est radicalement différente dans le cas des maladies dominantes, car le parent transmetteur sera lui-même malade, et que un foetus sur deux, statistiquement, sera porteur, conduisant en cela à une interruption de grossesse. L’expérience montre que les femmes sont plus demandeuses du test présymptomatique, les (futures) mères semblant plus concernées par le fait de devenir malades et de transmettre la maladie. Toutefois, une grossesse en cours ne conduit pas automatiquement à la demande d’un test présymptomatique [5]. Enfin, l’âge moyen de demande de diagnostic présymptomatique étant de 30 ans, la plupart des personnes accédant à un diagnostic présymptomatique ont déjà des enfants.
Si le nombre de demandes d’un test présymptomatique a eu tendance à augmenter après la découverte du gène impliqué dans la maladie de Huntington, les demandes de diagnostic prénatal après un test défavorable sont restées faibles : en effet, les résultats d’une étude européenne montrent que 85 % des sujets porteurs n’ont pas eu de grossesse, de même que 72 % des sujets non porteurs [10]. Parmi les porteurs qui ont eu des grossesses, 60 % ont demandé un diagnostic prénatal.
Autres maladies neurodégénératives pour lesquelles il existe un test présymptomatique
Les personnes à risque pour d’autres maladies neurodégénératives peuvent maintenant demander un test présymptomatique (Tableau I). Cependant, les gènes impliqués sont connus depuis moins longtemps, et certaines de ces pathologies sont très rares, d’où une expérience plus limitée.
Notre expérience concerne surtout les ataxies cérébelleuses autosomiques dominantes, causées par les mutations des gènes SCA (spinocerebellar ataxia) 1, 2, 3, 6 et 7. Certaines différences de comportements ont pu être mises en évidence avec les sujets demandant un test présymptomatique de la maladie de Huntington. En termes de motivations, les personnes à risque pour une ataxie cérébelleuse dominante mettent en avant leur crainte d’être atteint, rare chez les personnes à risque pour la maladie de Huntington (24 % versus 7 %). En revanche, le désir de savoir est plus grand chez les personnes à risque pour la maladie de Huntington (57 % versus 35 %), de même que le souhait d’informer leurs enfants (25 % versus 8 %) [6]. Cela souligne la difficulté supplémentaire des personnes à risque dans la maladie de Huntington, qui savent qu’elles ne se rendront pas compte du début de la maladie, en raison de l’anosognosie inhérente à l’affection, très différente d’un déni actif. Les personnes à risque pour une ataxie cérébelleuse dominante sont au contraire les premières à s’apercevoir du début des troubles. Le problème de la transmission à la descendance est par ailleurs plus important dans les SCA que dans la maladie de Huntington, d’où l’observation que les grossesses chez un couple porteur s’accompagnent beaucoup plus souvent d’une demande de diagnostic prénatal dans les SCA que dans la maladie de Huntington. Néanmoins, ces différences de motivations entre les groupes de personnes à risque n’empêchent pas la survenue, dans les deux pathologies, d’événements indésirables (dépression ou autres) après le rendu des résultats du test. Ces maladies bénéficient donc toutes d’une prise en charge pluridisciplinaire au long cours, indépendamment de leurs particularités propres en termes de manifestations (présence de troubles du comportement ou non) et de progression (perte d’autonomie ou décès rapide, versus maladie lentement progressive sans retentissement sur l’espérance de vie).
Les nouvelles pathologies accessibles à un test présymptomatique soulèveront d’autres interrogations : ainsi, alors que la pénétrance de la maladie de Huntington ou des SCA est complète, impliquant qu’être porteur signifie obligatoirement développer la maladie, la pénétrance est incomplète dans le cas des paraplégies spastiques autosomiques dominantes (SPG4, par exemple) ou avec certaines mutations de la maladie de Creutzfeldt-Jakob (E200K, par exemple) ; le fait de pouvoir encore échapper à la maladie après un résultat défavorable a forcément une influence sur la décision de faire ou non le test présymptomatique.
Conclusions
En France, la structure d’accueil pour le test présymptomatique de maladie de Huntington, mise en place dès 1992, permet aujourd’hui de mieux connaître les motivations et les souhaits des demandeurs, les conséquences de la prédiction sur leur vie et sur leur descendance, et l’intérêt d’une structure de prise en charge multidisciplinaire.
La consultation de diagnostic présymptomatique doit être réalisée par une équipe pluridisciplinaire accueillant le demandeur dans le respect de la temporalité de la prise de décision et d’une annonce difficile. La question n’est pas seulement « être (porteur) ou ne pas être (porteur) ». Elle serait plutôt : « comment être ». Éclairer une personne sur son statut génétique sans pouvoir lui proposer une prévention ou de traitement doit être mené sans précipitation.
Figure 2
Prise en charge multidisciplinaire de la demande de test prédictif.
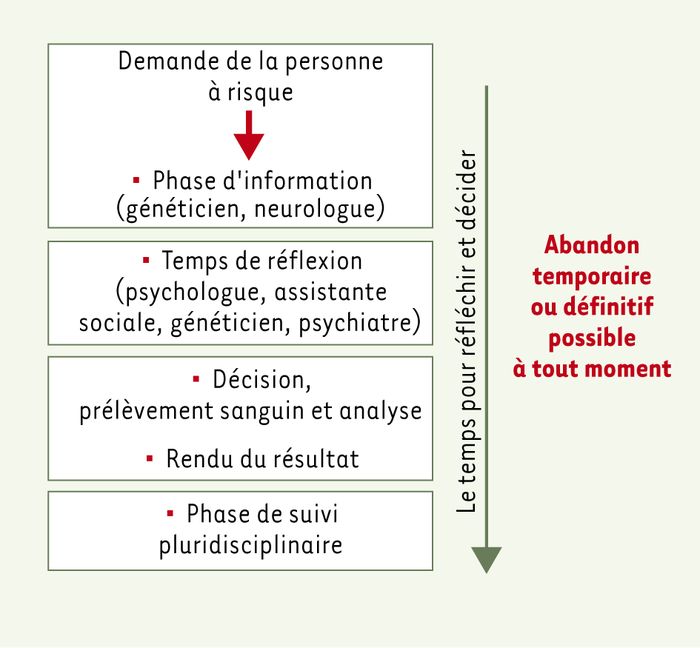
Commentaires (voir texte).
Parties annexes
Références
- 1. Bates G, Harper PS, Jones L. Huntington’s disease, 3rd ed. Oxford, UK : Oxford University Press, 2002 : 28-61.
- 2. Holmes SE, O’Hearn E, Rosenblatt A, et al. A repeat expansion in the gene encoding junctophilin-3 is associated with Huntington disease-like 2. Nat Genet 2001 ; 29 : 377-8.
- 3. Krause A, Temlett J, Van der Meyden K, et al. CAG/CTG repeat expansions at the HDL2 locus are a common cause of Huntington disease in Black South Africans. Am J Hum Genet 2002 ; 71 : 528.
- 4. International Huntington Association and World Federation of Neurology. Guidelines for the molecular genetics predictive tests in Huntington disease. Neurology 1994 ; 44 : 1533-6.
- 5. Lesca G, Goizet C, Durr A, and the French group for presymptomatic testing in neurogenetic disorders. Predictive testing in the context of pregnancy: experience in Huntington’s disease and autosomal dominant cerebellar ataxia. J Med Genet 2002 ; 39 : 522-5.
- 6. Goizet C, Lesca G, Durr A, and the French group for presymptomatic testing in neurogenetic disorders. Presymptomatic testing in Huntington’s disease and autosomal dominant cerebellar ataxias. Neurology 2002 ; 59 : 1330-6.
- 7. Almqvist EW, Bloch M, Brinkman R, et al. A worldwide assessment of the frequency of suicide, suicide attempts, or psychiatric hospitalization after predictive testing for Huntington disease. Am J Hum Genet 1999 ; 64 : 1293-304.
- 8. Gargiulo M. Guérir du risque ? Médecine prédictive. Quelle place pour l’homme ? Revue Laennec Médecine Santé Éthique 1999 ; 3-4 (n° spécial) : 16-9.
- 9. Evers-Kiebooms G, Zoeteweij MW, Harper P. Prenatal testing for late onset neurogenetic diseases. London: Bios Scientific Publishers Limited, 2002.
- 10. Evers-Kiebooms G, Nys K, Harper P, et al. Predictive DNA-testing for Huntington’s disease and reproductive decision making: a European collaborative study. Eur J Hum Genet 2002 ; 10 : 167-76.
Liste des figures
Figure 1
Bénéfice médical du diagnostic présymptomatique des maladies héréditaires.
Le bénéfice médical d’un diagnostic présymptomatique varie selon que l’on dispose ou non de méthodes de surveillance, de prévention ou de traitement pour la maladie (voir texte). MEN2 : néoplasies endocrines multiples de type 2.
Figure 2
Prise en charge multidisciplinaire de la demande de test prédictif.
Commentaires (voir texte).
Liste des tableaux
Tableau I
Exemples de maladies neurodégénératives pour lesquelles il existe des formes héréditaires et où un test prédictif est possible dans certains sous-groupes génétiques.