Corps de l’article
Il apparaît de plus en plus évident que la forme pathologique de la protéine du prion, la PrPSc, ne peut pas, à elle seule, rendre compte des phénomènes de neurodégénérescence observés dans les maladies à prion. Il a en effet été montré que cette isoforme, résistante à la protéinase K, n’était pas toujours détectée dans les encéphalopathies spongiformes transmissibles, en particulier dans les formes familiales humaines ou au cours de la transmission expérimentale de l’encéphalopathie spongiforme bovine. Par ailleurs, et de façon plus remarquable encore, l’accumulation de PrPSc dans le cerveau n’est pas toujours accompagnée d’une neurodégénérescence, comme le confirme un article récent [1]. En revanche, des formes intracytoplasmiques ou transmembranaires dérivées de la protéine cellulaire normale, la PrPC, se sont révélées toxiques pour les cellules neuronales aussi bien en culture cellulaire que dans des modèles animaux [2]. On peut ainsi se demander laquelle, de la forme cellulaire ou « scrapie », est la plus toxique ?
L’article récent d’A. Williamson et al. du Scripps Institute (La Jolla, CA, USA) [3], publié dans la revue Science, offre un éclairage intéressant sur ce problème. En effet, ces auteurs ont pu montrer que des anticorps bivalents dirigés contre la PrPC induisaient, lorsqu’ils étaient injectés par voie stéréotaxique dans l’hippocampe de souris, une apoptose et des dommages neuronaux étendus au point de l’injection. Des anticorps sans relation avec la PrPC, ainsi que des anticorps anti-PrPC monovalents ou déjà saturés avec la protéine prion ne provoquent pas de lésions similaires. On peut donc établir un modèle dans lequel la neurodégénérescence serait liée à la perturbation ou au détournement de l’action physiologique de la PrPC, action dépendante d’une dimérisation de la molécule (Figure 1). Il n’est pas anodin en effet que la fonction physiologique de la protéine du prion se rapporte - dans différents modèles expérimentaux - à la survie neuronale et à la défense contre le stress oxydant, par l’intermédiaire de différentes voies de signalisation cellulaire.
Figure 1
La fonction physiologique de la protéine du prion mise en évidence dans différents modèles est corrélée à la survie neuronale et à la défense contre le stress oxydant.
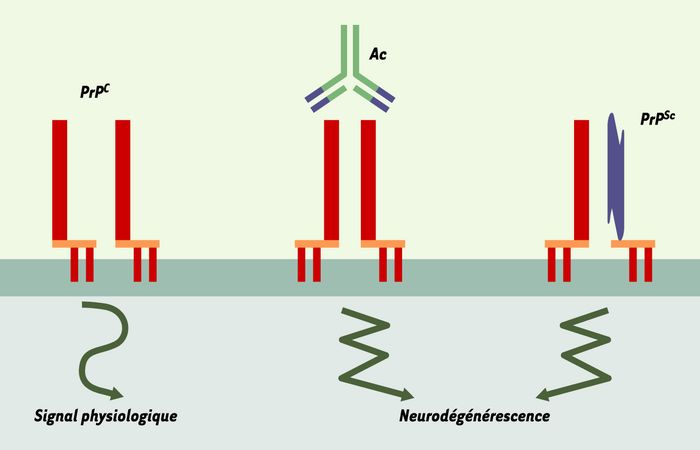
Cette fonction pourrait être liée à la formation d’un complexe de signalisation associant une ou plusieurs (dimères) molécules de PrPC et d’autres partenaires (récepteur de la laminine) non représentés ici. Le pontage in vivo de molécules de PrPC par des anticorps (Ac) [3] ou la présence concomitante de PrPC et de PrPSc au niveau de la membrane [6] pourraient entraîner l’altération du signal physiologique de la PrPC, conduisant à un processus d’apoptose neuronale et de neurodégénérescence.
Toutefois, au vu de la littérature récente, un certain nombre de questions se posent. Une première est d’ordre thérapeutique : différentes études indépendantes ont montré une abolition de la production de PrPScin vitro et in vivo en présence d’anticorps dirigés contre la PrP [4] ; dans ces études, cependant, aucun signe de toxicité n’a pu être observé, un résultat en désaccord avec les conclusions et les craintes de L. Solforosi et al. [3]. Certes, il ne s’agissait ni de la même voie d’administration, ni des mêmes anticorps que ceux utilisés par L. Solorosi, mais certains reconnaissaient les mêmes épitopes. Un autre aspect de ces résultats est troublant et concerne les études de la fonction physiologique de la PrPC. En effet, plusieurs auteurs ont parfois utilisé pour ces études des anticorps bivalents et montré que la PrP participait à des voies de signalisation qui, loin d’être toxiques, favoriseraient au contraire la survie neuronale [5]. Comment réconcilier ces résultats avec ceux de L. Solforosi et al. ? Le fait que les épitopes ciblés soient différents suggère que des régions différentes de la PrP puissent participer à des fonctions différentes. Certains épitopes/anticorps pourraient favoriser le « blocage » fonctionnel de la PrPC, et d’autres plutôt son activation. Enfin, on ne peut s’empêcher de penser que des résultats fonctionnels obtenus par des méthodes d’études uniquement fondées sur l’utilisation d’anticorps et/ou ne prenant en compte qu’un nombre limité d’épitopes de la protéine doivent être confirmés par d’autres approches.
En conclusion, l’étude du groupe de A. Williamson est une contribution importante aux recherches sur les prions, qui alimente un débat complexe en suggérant que la forme normale de la PrP participe à la toxicité, et appelle à la prudence quant à l’utilisation d’anticorps dirigés contre cette protéine dans des perspectives thérapeutiques.
Parties annexes
Références
- 1. Mallucci G, Dickinson A, Linehan J, et al. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science 2003 ; 302 : 871-4.
- 2. Chiesa R, Harris DA. Prion diseases : what is the neurotoxic molecule ? Neurobiol Dis 2001 ; 8 : 743-63.
- 3. Solforosi L, Criado JR, McGavern DB, et al. Cross-linking cellular prion protein triggers neuronal apoptosis in vivo. Science 2004 ; 303 : 1514-6.
- 4. White AR, Enever P, Tayebi M, et al. Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature 2003 ; 422 : 80-3.
- 5. Mouillet-Richard S, Ermonval M, Chebassier C, et al. Signal transduction through prion protein. Science 2000 ; 289 : 1925-8.
- 6. Lehmann S, Harris DA. Mutant and infectious prion proteins display common biochemical properties in cultured cells. J Biol Chem 1996 ; 271 : 1633-7.
Liste des figures
Figure 1
La fonction physiologique de la protéine du prion mise en évidence dans différents modèles est corrélée à la survie neuronale et à la défense contre le stress oxydant.
Cette fonction pourrait être liée à la formation d’un complexe de signalisation associant une ou plusieurs (dimères) molécules de PrPC et d’autres partenaires (récepteur de la laminine) non représentés ici. Le pontage in vivo de molécules de PrPC par des anticorps (Ac) [3] ou la présence concomitante de PrPC et de PrPSc au niveau de la membrane [6] pourraient entraîner l’altération du signal physiologique de la PrPC, conduisant à un processus d’apoptose neuronale et de neurodégénérescence.