Résumés
Résumé
Le ciblage de l’anomalie génétique à l’origine d’une maladie constitue le nouveau défi de la recherche pharmaceutique. Précurseur en ce domaine, l’imatinib mésylate (Glivec®, Novartis) représente à l’heure actuelle une révolution dans le traitement de la leucémie myéloïde chronique, car cette molécule inhibe l’activité tyrosine kinase de l’oncogène c-abl. Bien que minoritaire, la résistance clinique au traitement de la leucémie myéloïde chronique par l’imatinib mésylate a cependant suscité bien des investigations, permettant l’acquisition de nombreuses données fondamentales concernant le mode d’action des tyrosine kinases et certains mécanismes moléculaires associés à l’évolution de la maladie. Par ailleurs, la compréhension des bases moléculaires de cette résistance a permis de montrer que la spécificité de l’imatinib mésylate présente des limites, l’utilisation prolongée de ce médicament chez des patients porteurs de certaines mutations ponctuelles de c-abl pouvant précipiter l’évolution de la maladie. Cet article expose comment l’histoire de l’utilisation de l’imatinib mésylate représente un exemple de parfaite synergie entre l’expérimentation clinique et la recherche fondamentale
Summary
For drug development and pharmaceutical research, targeting the molecular abnormalities is considered as a new challenge. A number of diseases including cancer are linked to perturbation of tyrosine kinase (TK). Imatinib (Glivec® or Gleevec®, Novartis), the most potent inhibitor of c-abl TK, was recently developed. This molecule has been approved in the treatment of chronic myeloid leukemia (CML). However, emergence of clinical resistance regarding a low rate of CML patients leads to intensive research. In the current article, we discuss the data and the mechanism of the resistance phenomenon. This review illustrates the important requirement to transfer back the information from patient to laboratory in order to improve future drug design.
Corps de l’article
Les protéine kinases constituent d’importants éléments régulateurs de la prolifération cellulaire, de la différenciation et de l’apoptose. Elles jouent un rôle majeur dans la cascade d’événements observés lors de la transduction d’un signal. Durant le développement embryonnaire et la vie adulte, les mécanismes de signalisation cellulaire doivent être coordonnés et intégrés. Tout événement rompant cet équilibre peut conduire à la formation d’une tumeur maligne. Ainsi, le développement d’inhibiteurs sélectifs de certaines protéine kinases présentant un dysfonctionnement constitue une approche thérapeutique prometteuse dans le contrôle de certains cancers.
Dans ce contexte, la tyrosine kinase (TK) abelson (c-abl) représente une cible thérapeutique de choix. En effet, il a été démontré qu’une activation permanente de c-abl est à l’origine du processus de leucémogenèse dans la leucémie myéloïde chronique (LMC) [1] (voir Encadré). Parmi les différents dérivés testés de la 2-phénylaminopyrimidine, le STI 571 (signal transduction inhibitor 571), ou imatinib mésylate (IM), a démontré une capacité d’inhibition importante sur l’activité TK de la protéine de fusion bcr-abl [2, 3]. La connaissance structurale du site actif de la TK c-abl a permis de mieux comprendre le mécanisme d’action de l’IM [4-6] (Figure 1). Contrairement à ce qui était encore récemment envisagé, cette molécule n’agit pas seulement par compétition directe avec les molécules d’ATP, mais, surtout, elle stabilise la forme inactive de c-abl en déplaçant l’équilibre physiologique de conformation au détriment de la forme active. Commercialisée sous le nom de Glivec®, cette molécule sélective représente un espoir considérable dans le traitement de la LMC.
Figure 1
Représentation tridimensionnelle du site actif de c-abl.

Trois boucles fonctionnelles ont été modélisées : une boucle C, correspondant au site catalytique de la TK , une boucle P (phosphate binding loop), qui structure le site de fixation de l’ATP et dans laquelle se lie l’IM, et une boucle d’activation A, dont la conformation dépend de son statut de phosphorylation. Le domaine très conservé aminoterminal de la protéine représente un élément essentiel dans l’auto-inhibition de la TK. Cette région, très riche en hélices α, interagit avec la boucle A et permet sa stabilisation sous forme active. A. Equilibre physiologique entre la forme active de la molécule et la forme inactive. Lorsque la boucle A présente un statut phosphorylé, sa conformation « ouverte » stable permet une grande accessibilité du site catalytique aux molécules d’ATP. La TK c-abl est alors sous forme active. En revanche, lorsque la boucle A présente un statut déphosphorylé, sa conformation spécifique rend l’accès du site catalytique aux substrats difficile. La protéine c-abl présente alors une conformation inactive qui n’est pas similaire à celles que l’on peut observer pour d’autres TK. B. Interaction entre une molécule d’imatinib mésylate (IM) et c-abl. La conformation de la boucle A joue un rôle majeur dans l’affinité de la TK pour son substrat. Elle est le siège de la spécificité de l’IM, car cette dernière reconnaît la conformation inactive très particulière de c-abl.
La LMC comme modèle d’étude
Cette maladie représente un modèle privilégié d’étude d’inhibiteurs de TK, car l’efficacité du traitement est évaluée simplement sur le nombre de globules blancs (dont la normalité évoque une rémission hématologique) et de cellules leucémiques porteuses du chromosome Philadelphie (chr Ph) ou de son équivalent moléculaire, le transcrit de fusion bcr-abl. Une majorité de patients (95 %) atteints de LMC, traités par l’IM en phase chronique de la maladie, présentent une rémission hématologique. Cette rémission est associée dans 85 % des cas à une réponse cytogénétique majeure ou complète (perte du chr Ph). En revanche, on ne comprend pas pourquoi l’IM perd de son efficacité lors de l’évolution de la maladie (seulement 29 % des patients traités par l’IM en phase accélérée présentent une rémission hématologique) [7] ((→) m/s 2003, n° 6-7, p. 667), ni pourquoi certains patients rechutent après une bonne réponse initiale au traitement.
Étude moléculaire des mécanismes de résistance
L’étude des mécanismes de résistance a été menée principalement sur des lignées cellulaires rendues résistantes par exposition à des doses croissantes d’IM. L’hétérogénéité des lignées utilisées a parfois donné des résultats contradictoires. Cette approche in vitro a été complétée par des travaux réalisés sur des cellules sanguines ou médullaires de patients en phase accélérée de LMC et n’ayant jamais présenté de réponse hématologique favorable au traitement, et sur des cellules de patients atteints de leucémie aiguë lymphoblastique (LAL) associée à la présence d’un chr Ph (LAL Ph+). Ces approches ont permis d’élucider certains mécanismes de résistance.
Mécanismes de résistance hors mutations
Réactivation de l’activité TK de bcr-abl
Une modification quantitative de la cible par amplification du gène bcr-abl peut être à l’origine d’une réactivation de l’activité TK dans les cellules devenues résistantes à l’IM [8] ((→) m/s 2001, n° 11, p. 1192).
Cependant, le phénotype résistant observé est réversible puisque la suppression de l’IM du milieu de culture conduit à la diminution du taux de synthèse de l’ARNm bcr-abl et de la protéine chimérique. Ce mécanisme de résistance est observé chez moins de 5 % des patients ayant eu une recherche d’amplification génique [8-12].
Le gène MDR-1
Il a été démontré dans de nombreux cancers (comme les leucémies aiguës myéloblastiques de l’adulte) qu’une augmentation de la transcription du gène MDR1 (multidrug resistance), déterminant la synthèse de la pompe d’efflux de la glycoprotéine P, était directement corrélée à la résistance. Dans la résistance à l’IM, une augmentation conjointe de l’expression de bcr-abl et de MDR1 contribue à la résistance d’une lignée LAMA84R [13]. Dans un second modèle issu d’une lignée érythroïde K562, la résistance à la doxorubicine (un agent intercalant de l’ADN) dépendante de MDR1 protège la cellule contre l’action de l’IM [13]. Enfin, l’augmentation de l’expression de MDR1 induite par transfection contribue à la résistance d’une lignée AR230 à de faibles doses d’IM [8], mais n’a aucun effet sur une autre lignée K562 [14].
In vivo, dans la LMC, la résistance à l’IM liée à MDR1 est difficile à évaluer en raison d’une expression physiologique importante de ce gène dans les cellules sanguines. Notons cependant l’observation d’une surexpression de MDR1 au cours de l’évolution de LAL Ph+ résistantes à l’IM [15].
L’accessibilité intracellulaire de l’IM
L’IM a la capacité de se fixer sur une glycoprotéine acide α-1 plasmatique (AGP). De manière intéressante, la survie d’animaux transgéniques exprimant bcr-abl et résistants à l’IM est améliorée si la prise de ce médicament est associée à celle de l’érythromycine. Dans ce modèle, la compétition par l’érythromycine pour la fixation à l’AGP a permis d’accroître l’efficacité du traitement par IM [16].
In vivo, la corrélation entre résistance à l’IM et taux d’AGP n’a pas toujours été confirmée [17-20]. Il est seulement possible de montrer une corrélation entre un taux élevé d’AGP et la résistance à l’IM chez des patients en phase blastique de la maladie [17, 20].
L’activation d’autres voies de survie
Pour une concentration d’IM donnée, un délai de cent heures est nécessaire pour obtenir l’apoptose in vitro de cellules leucémiques de patients atteints de LMC, alors que l’apoptose de cellules blastiques de patients atteints de LAL Ph+ est acquise en 16 à 20 heures [21]. Cette cinétique d’induction d’apoptose moins rapide pour les cellules de LMC pourrait résulter de l’activation accrue d’autres voies de survie, indépendantes de bcr-abl. On peut supposer que dans les cellules de LAL Ph+, ces autres voies puissent être affectées par des anomalies génétiques additionnelles.
Un modèle de cellules K562, résistantes à l’IM, mais présentant une diminution du taux d’expression du transcrit bcr-abl, a permis de montrer que la résistance pouvait résulter de l’activation de la voie des src-kinases [22]. L’augmentation de la phosphorylation de molécules telles que LYN, responsables de la signalisation en aval, permettrait de contourner l’inhibition de bcr-abl par l’IM dans ce modèle. In vivo, l’augmentation de l’activité de la src-kinase LYN, observée chez certains patients en phase accélérée de LMC, est probablement liée à l’évolution de la maladie, mais pourrait participer à la résistance à l’IM [22].
Résistance par mutations
Initialement, une équipe américaine a montré la présence d’une mutation ponctuelle dans le domaine TK de c-abl aboutissant à la substitution d’une thréonine par une isoleucine en position 315 (T315I) chez six patients résistants au traitement par IM [9]. Dans le même temps, deux équipes européennes ont décrit la présence d’autres mutations ponctuelles affectant un acide glutamique (E255) situé dans la boucle P du domaine kinase chez des patients ayant rechuté après une réponse initiale au traitement [12, 23]. À l’heure actuelle, on dénombre 22 mutations affectant 19 acides aminés différents [23-26], avec une fréquence très variable d’une étude à l’autre. Cette variabilité dépend beaucoup de la méthodologie utilisée. Par ailleurs, ces études portent sur des patients à des stades d’évolution différents de leur LMC. Or, la fréquence des mutations observées est plus importante chez des patients ayant rechuté sous IM après une réponse cytogénétique initiale favorable que chez des patients ne l’ayant pas présentée. Ainsi, une étude récemment publiée démontre que la fréquence des mutations détectées par séquençage augmente considérablement chez des patients en phase accélérée, dont le diagnostic de LMC a été posé quatre ans au moins avant la date de la recherche de mutations (27 patients porteurs de mutations sur 104 étudiés, quatre ans après le diagnostic, contre 0 sur 40 patients étudiés dont le diagnostic date de moins de quatre ans) [24].
In vitro, une approche de mutagenèse aléatoire a permis de constituer une banque de clones cellulaires BaF3 porteurs de mutations ponctuelles du gène bcr-abl [27]. Les clones analysés ont été sélectionnés pour leur résistance à l’IM. Cette approche originale a permis de retrouver toutes les mutations initialement décrites dans le site actif de c-abl chez les patients résistants au traitement. Ce travail a également permis de caractériser d’autres mutations situées sur la face opposée du domaine kinase de la molécule, ou dans la région aminoterminale de c-abl. L’étude structurale de ces variants a permis de mettre en évidence une nouvelle catégorie de mutations capables de déstabiliser la conformation inactive de la TK par effet allostérique.
Importance des mutations dans la résistance au traitement
L’effet des mutations de c-abl sur la résistance à l’IM varie selon leur nature ou leur localisation. Dans la LMC, les mutations situées dans la boucle P présentent une valeur pronostique défavorable chez les patients. En effet, dans une étude rétrospective, S. Brandford et al. ont montré que 92 % des patients traités par IM, ayant une ou plusieurs mutations dans cette région, décèdent dans les six mois qui suivent leur détection, alors que si les mutations sont situées dans une autre région du site actif de c-abl, la proportion de décès est de 21 % [24]. L’analyse structurale de la boucle P montre qu’une mutation affectant l’acide aminé 253 ou 255 modifie la conformation tridimensionnelle, perturbant l’interaction de l’IM avec le site catalytique de c-abl [28]. De plus, la substitution d’une tyrosine par une phénylalanine en position 253 confère à la TK une activité oncogénique accrue, puisque les cellules bcr-abl portant cette mutation sont capables de produire une quantité plus importante de phosphotyrosines in vitro [29]. Il est donc tout à fait possible que ce type de mutation détermine non seulement une résistance importante à l’IM, mais contribue également à l’accélération de la maladie.
Les mutations localisées dans la boucle A, comme la substitution de l’histidine en position 396, influent sur l’affinité de l’IM pour son substrat. De même, la substitution des résidus hydrophobes en position 351 ou 486 perturbe l’interaction avec la région aminoterminale de la TK. Ainsi, ces mutations déstabilisent la forme inactive de c-abl et déplacent l’équilibre vers sa configuration active, non reconnue par l’IM. L’analyse cellulaire et biochimique de ces mutations démontre que l’IM a une faible capacité d’inhibition de la prolifération cellulaire (IC50 de 4 à 9 μM)[29]. Cliniquement, on peut envisager qu’une augmentation de la posologie d’IM puisse améliorer la réponse au traitement chez certains patients.
Parmi les différentes substitutions altérant les points de liaison de l’IM dans le site actif de c-abl, la mutation T315I est associée à une importante réduction de la sensibilité des cellules à ce produit (IC50 ≥ 20 μM), car elle abolit une liaison hydrogène entre les deux partenaires [9]. Cette mutation est directement impliquée dans la résistance des patients au traitement et représente près de 60 % des mutations retrouvées.
D’où proviennent les mutations ?
Une constatation surprenante avait été rapportée par C. Barthe et al. chez un patient atteint d’une LMC et résistant à l’IM [30] : la mutation de l’acide aminé 255, détectée 13 mois après la mise en route du traitement, n’était pas retrouvée au début de la prise d’IM. L’augmentation du signal de détection de cette mutation par séquençage au cours du temps témoignait toutefois d’un enrichissement de la population de cellules mutées au cours du traitement et pouvait légitimement suggérer un effet mutagène de l’IM in vivo. Cependant, l’exposition des lignées cellulaires à de très fortes doses d’IM n’entraîne pas l’émergence de mutations. Par ailleurs, des mutations ont été détectées chez des patients présentant une résistance après moins de trois mois de traitement, délai trop court pour que l’IM puisse induire des mutations.
Par le développement d’une technique de détection de mutations plus sensible que le séquençage, nous avons pu démontrer, par PCR spécifique d’allèle, la présence de rares cellules mutées (1/10 000), porteuses de la mutation T315I, avant la mise en route du traitement par l’IM chez certains patients ayant reçu de l’interféron, mais également au moment du diagnostic chez un patient (Figure 2) [26, 31]. La présence des mutations T315I et E255K avant la prise d’IM a été confirmée dans deux autres études, par séquençage rétrospectif de prélèvements de patients chez lesquels le traitement n’avait pas entraîné de réponse hématologique. Ces différents résultats ont permis de suggérer que l’émergence de la résistance à l’IM pouvait être expliquée par la préexistence de cellules bcr-abl mutées dans le site de la TK, dont le faible nombre rendait toutefois impossible la détection initiale par séquençage. Il est ensuite possible que la résistance à l’IM de ces cellules leur confère un avantage sélectif sous traitement. Ainsi, suivant le moment d’apparition des mutations, on peut envisager les scénario illustrés dans la Figure 3.
Figure 2
Détection de la mutation T315I par PCR spécifique d’allèle (PCR-ASO).
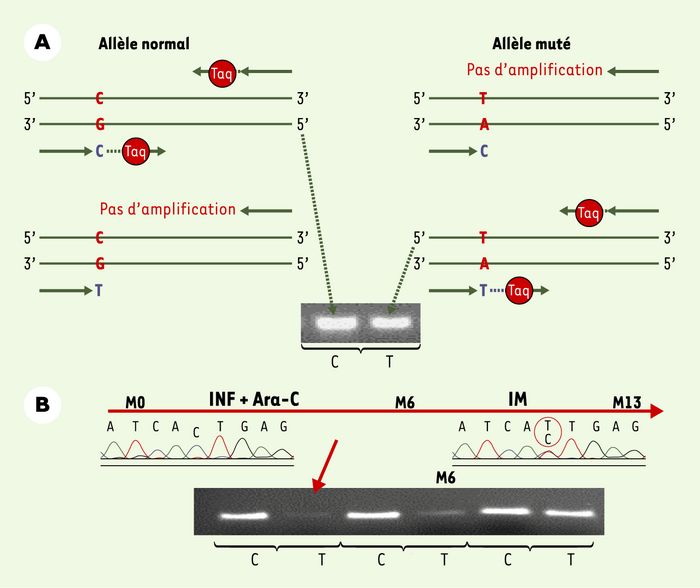
A. Principe de la PCR-ASO. Cette technique est fondée sur la spécificité de séquence des amorces utilisées pour la réaction de PCR. Pour chaque analyse, la réaction est réalisée en double de manière simultanée, sur l’ADN du patient, en utilisant une amorce « anti-sens » commune et une amorce « sens » dont la séquence terminale est spécifique de la mutation ou de l’allèle normal. En cas de non appariement, la réaction d’élongation par la Taq polymérase ne peut se faire. Ainsi, l’analyse des produits de la réaction après électrophorèse en gel d’agarose permet de visualiser la présence ou l’absence d’un produit d’amplification suivant le couple d’amorces utilisé, permettant ainsi de déterminer la présence ou l’absence, dans l’échantillon d’ADN, de l’allèle muté. B. Cinétique d’évolution d’un clone porteur de la mutation T315I dans le cas de la prise en charge d’un patient atteint de LMC. M0 : diagnostic et mise en route du traitement par interféron et aracytine (INF+Ara-C) ; M6 : mise en route du traitement par IM (six mois après le diagnostic) ; M13 : absence de réponse cytogénétique après 7 mois de traitement par IM. Le diagramme de séquençage ne détecte pas de mutation à M0 mais détecte une double population (séquence normale C et séquence mutée T) à M13. La cinétique d’évolution par PCR-ASO des cellules mutées témoigne chez ce patient d’un enrichissement du clone muté (minoritaire au diagnostic et non détecté par séquençage) sous traitement par IM.
Figure 3
Cinétique d’émergence dans le sang des différentes populations cellulaires.

Au cours de la progression de la phase chronique de la maladie vers la phase d’accélération, des mutations somatiques s’accumulent dans les cellules cancéreuses (partie supérieure de la figure). Certaines mutations sont délétères. D’autres n’altèrent pas l’intégrité cellulaire, car elles n’affectent pas la fonction de la TK de c-abl, et sont même capables d’apporter un avantage sélectif à la cellule (comme la mutation T253F). Ces événements aléatoires seraient passés inaperçus si un facteur extrinsèque, l’imatinib mésylate (IM), ne leur avait pas attribué un avantage sélectif considérable. La prolifération clonale de ces cellules résistantes au traitement est favorisée sous IM par rapport à celle des autres clones malins. C’est pourquoi, suivant le moment d’apparition de la mutation au cours du traitement, il est possible d’observer une rémission complète, une résistance précoce, ou une rechute après une réponse initiale.
Conclusions et perspectives
Ces études démontrent les limites de la spécificité d’action de l’IM. Une mutation ponctuelle, n’affectant ni la fonction de la TK, ni la viabilité de la cellule, suffit à abolir ou à restreindre l’action de ce médicament. Or, le ciblage des anomalies moléculaires responsables de certaines maladies représente, à l’heure actuelle, le nouveau défi de la recherche pharmaceutique. Cette recherche risque d’être confrontée à l’instabilité du génome humain, puisque le profil de réponse à une molécule pourrait être déterminé par la variabilité génétique de l’hôte.
On peut établir une analogie entre ce que la médecine a connu avec l’avènement des antibiotiques ou des antiviraux, et l’espoir que représente l’IM pour le traitement des LMC et de certains autres cancers, pour lesquels on ne disposait, jusqu’à ce jour, d’aucune ressource thérapeutique. Nous savons que l’efficacité spectaculaire des antibiotiques a été contrecarrée par les variations génétiques des bactéries, à l’origine de résistances. De même, l’efficacité de l’AZT (azidothymidine) dans le traitement du sida a été remise en question lorsque des mutations dans la séquence de la transcriptase inverse du VIH a entraîné une résistance au traitement. Il a fallu alors développer de nouveaux médicaments et associer plusieurs molécules inhibitrices pour surmonter ces résistances.
Des essais thérapeutiques sont entrepris actuellement pour évaluer l’efficacité de l’association de l’IM avec des médicaments ciblant une autre voie de prolifération cellulaire. Par ailleurs, des dérivés de l’IM ne présentant pas certaines contraintes structurales de c-abl (comme la boucle A) sont étudiés en laboratoire [32]. Il est certain que le développement de l’IM va s’inscrire dans une longue continuité d’efforts fructueux pour le développement des médicaments de demain.
Parties annexes
Références
- 1. Shtivelman E, Lifshitz B, Gale RP, et al. Fused transcript of abl and bcr genes in chronic myelogenous leukaemia. Nature 1985 ; 315 : 550-4.
- 2. Buchdunger E, Zimmermann J, Mett H, et al. Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative. Cancer Res 1996 ; 56 : 100-4.
- 3. Druker BJ, Lydon NB. Lessons learned from the development of an abl tyrosine kinase inhibitor for chronic myelogenous leukemia. J Clin Invest 2000 ; 105 : 3-7.
- 4. Schindler T, Bornmann W, Pellicena P, et al. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science 2000 ; 289 : 1938-42.
- 5. Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell 2002 ; 109 : 275-82.
- 6. Dorey K, Engen JR, Kretzschmar J, et al. Phosphorylation and structure-based functional studies reveal a positive and a negative role for the activation loop of the c-Abl tyrosine kinase. Oncogene 2001 ; 20 : 8075-84.
- 7. O’Brien SG, Guilhot F, Larson RA, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med 2003 ; 348 : 994-1004.
- 8. Mahon FX, Deininger MW, Schultheis B, et al. Selection and characterization of BCR-ABL positive cell lines with differential sensitivity to the tyrosine kinase inhibitor STI571 : diverse mechanisms of resistance. Blood 2000 ; 96 : 1070-9.
- 9. Gorre ME, Mohammed M, Ellwood K, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 2001 ; 293 : 876-80.
- 10. Le Coutre P, Tassi E, Varella-Garcia M, et al. Induction of resistance to the Abelson inhibitor STI571 in human leukemic cells through gene amplification. Blood 2000 ; 95 : 1758-66.
- 11. Hochhaus A, Kreil S, Corbin AS, et al. Molecular and chromosomal mechanisms of resistance to imatinib (STI571) therapy. Leukemia 2002 ; 16 : 2190-6.
- 12. Hochhaus A, Kreil S, Corbin A, et al. Roots of clinical resistance to STI-571 cancer therapy. Science 2001 ; 293 : 2163.
- 13. Mahon FX, Belloc F, Lagarde V, et al. MDR1 gene overexpression confers resistance to imatinib mesylate in leukemia cell line models. Blood 2003 ; 101 : 2368-73.
- 14. Ferrao PT, Frost MJ, Siah SP, et al. Overexpression of P-glycoprotein in K562 cells does not confer resistance to the growth inhibitory effects of Imatinib (STI571) in vitro. Blood 2003 ; 102 : 4499-503.
- 15. Hofmann WK, de Vos S, Elashoff D, et al. Relation between resistance of Philadelphia-chromosome-positive acute lymphoblastic leukaemia to the tyrosine kinase inhibitor STI571 and gene-expression profiles : a gene-expression study. Lancet 2002 ; 359 : 481-6.
- 16. Gambacorti-Passerini C, Barni R, le Coutre P, et al. Role of alpha1 acid glycoprotein in the in vivo resistance of human BCR-ABL(+) leukemic cells to the abl inhibitor STI571. J Natl Cancer Inst 2000 ; 92 : 1641-50.
- 17. Jorgensen HG, Elliott MA, Allan EK, et al. Alpha1-acid glycoprotein expressed in the plasma of chronic myeloid leukemia patients does not mediate significant in vitro resistance to STI571. Blood 2002 ; 99 : 713-5.
- 18. Gambacorti-Passerini C, Zucchetti M, Russo D, et al. Alpha1 acid glycoprotein binds to imatinib (STI571) and substantially alters its pharmacokinetics in chronic myeloid leukemia patients. Clin Cancer Res 2003 ; 9 : 625-32.
- 19. Le Coutre P, Kreuzer KA, Na IK, et al. Determination of alpha-1 acid glycoprotein in patients with Ph+ chronic myeloid leukemia during the first 13 weeks of therapy with STI571. Blood Cells Mol Dis 2002 ; 28 : 75-85.
- 20. Larghero J, Leguay T, Mourah S, et al. Relationship between elevated levels of the alpha 1 acid glycoprotein in chronic myelogenous leukemia in blast crisis and pharmacological resistance to imatinib (Gleevec) in vitro and in vivo. Biochem Pharmacol 2003 ; 66 : 1907-13.
- 21. Gambarcorti-Passerini C, Gunby R, Piazza R, et al. Molecular mechanisms of resistance to imatinib in Philadelphia-chromosome-positive leukaemias. Lancet Oncol 2003 ; 4 : 75-85.
- 22. Donato NJ, Wu JY, Stapley J, et al. BCR-ABL independence and LYN kinase overexpression in chronic myelogenous leukemia cells selected for resistance to STI571. Blood 2003 ; 101 : 690-8.
- 23. Barthe C, Cony-Makhoul P, Melo JV, et al. Roots of clinical resistance to STI-571 cancer therapy. Science 2001 ; 293 : 2163.
- 24. Branford S, Rudzki Z, Walsh S, et al. Detection of BCR-ABL mutations in patients with CML treated with imatinib is virtually always accompanied by clinical resistance, and mutations in the ATP phosphate-binding loop (P-loop) are associated with a poor prognosis. Blood 2003 ; 102 : 276-83.
- 25. Shah NP, Nicoll JM, Nagar B, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell 2002 ; 2 : 117-25.
- 26. Roche-Lestienne C, Soenen-Cornu V, Grardel-Duflos N, et al. Several types of mutations of the Abl gene can be found in chronic myeloid leukemia patients resistant to STI571, and they can pre-exist to the onset of treatment. Blood 2002 ; 100 : 1014-8.
- 27. Azam M, Latek RR, Daley GQ. Mechanisms of autoinhibition and STI-571/Imatinib resistance revealed by mutagenesis of bcr-abl. Cell 2003 ; 112 : 831-43.
- 28. Corbin AS, La Rosee P, Stoffregen EP, et al. Several Bcr-Abl kinase domain mutants associated with imatinib mesylate resistance remain sensitive to imatinib. Blood 2003 ; 101 : 4611-4.
- 29. Roumiantsev S, Shah NP, Gorre ME, et al. Clinical resistance to the kinase inhibitor STI-571 in chronic myeloid leukemia by mutation of Tyr-253 in the Abl kinase domain P-loop. Proc Natl Acad Sci USA 2002 ; 99 : 10700-5.
- 30. Barthe C, Gharbi MJ, Lagarde V, et al. Mutation in the ATP-binding site of BCR-ABL in a patient with chronic myeloid leukaemia with increasing resistance to STI571. Br J Haematol 2002 ; 119 : 109-11.
- 31. Roche-Lestienne C, Lai JL, Darre S, et al. A mutation conferring resistance to imatinib at the time of diagnosis of chronic myelogenous leukemia. N Engl J Med 2003 ; 348 : 2265-6.
- 32. Nagar B, Bornmann WG, Pellicena P, et al. Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571). Cancer Res 2002 ; 62 : 4236-43.
Liste des figures
Figure 1
Représentation tridimensionnelle du site actif de c-abl.
Trois boucles fonctionnelles ont été modélisées : une boucle C, correspondant au site catalytique de la TK , une boucle P (phosphate binding loop), qui structure le site de fixation de l’ATP et dans laquelle se lie l’IM, et une boucle d’activation A, dont la conformation dépend de son statut de phosphorylation. Le domaine très conservé aminoterminal de la protéine représente un élément essentiel dans l’auto-inhibition de la TK. Cette région, très riche en hélices α, interagit avec la boucle A et permet sa stabilisation sous forme active. A. Equilibre physiologique entre la forme active de la molécule et la forme inactive. Lorsque la boucle A présente un statut phosphorylé, sa conformation « ouverte » stable permet une grande accessibilité du site catalytique aux molécules d’ATP. La TK c-abl est alors sous forme active. En revanche, lorsque la boucle A présente un statut déphosphorylé, sa conformation spécifique rend l’accès du site catalytique aux substrats difficile. La protéine c-abl présente alors une conformation inactive qui n’est pas similaire à celles que l’on peut observer pour d’autres TK. B. Interaction entre une molécule d’imatinib mésylate (IM) et c-abl. La conformation de la boucle A joue un rôle majeur dans l’affinité de la TK pour son substrat. Elle est le siège de la spécificité de l’IM, car cette dernière reconnaît la conformation inactive très particulière de c-abl.
Figure 2
Détection de la mutation T315I par PCR spécifique d’allèle (PCR-ASO).
A. Principe de la PCR-ASO. Cette technique est fondée sur la spécificité de séquence des amorces utilisées pour la réaction de PCR. Pour chaque analyse, la réaction est réalisée en double de manière simultanée, sur l’ADN du patient, en utilisant une amorce « anti-sens » commune et une amorce « sens » dont la séquence terminale est spécifique de la mutation ou de l’allèle normal. En cas de non appariement, la réaction d’élongation par la Taq polymérase ne peut se faire. Ainsi, l’analyse des produits de la réaction après électrophorèse en gel d’agarose permet de visualiser la présence ou l’absence d’un produit d’amplification suivant le couple d’amorces utilisé, permettant ainsi de déterminer la présence ou l’absence, dans l’échantillon d’ADN, de l’allèle muté. B. Cinétique d’évolution d’un clone porteur de la mutation T315I dans le cas de la prise en charge d’un patient atteint de LMC. M0 : diagnostic et mise en route du traitement par interféron et aracytine (INF+Ara-C) ; M6 : mise en route du traitement par IM (six mois après le diagnostic) ; M13 : absence de réponse cytogénétique après 7 mois de traitement par IM. Le diagramme de séquençage ne détecte pas de mutation à M0 mais détecte une double population (séquence normale C et séquence mutée T) à M13. La cinétique d’évolution par PCR-ASO des cellules mutées témoigne chez ce patient d’un enrichissement du clone muté (minoritaire au diagnostic et non détecté par séquençage) sous traitement par IM.
Figure 3
Cinétique d’émergence dans le sang des différentes populations cellulaires.
Au cours de la progression de la phase chronique de la maladie vers la phase d’accélération, des mutations somatiques s’accumulent dans les cellules cancéreuses (partie supérieure de la figure). Certaines mutations sont délétères. D’autres n’altèrent pas l’intégrité cellulaire, car elles n’affectent pas la fonction de la TK de c-abl, et sont même capables d’apporter un avantage sélectif à la cellule (comme la mutation T253F). Ces événements aléatoires seraient passés inaperçus si un facteur extrinsèque, l’imatinib mésylate (IM), ne leur avait pas attribué un avantage sélectif considérable. La prolifération clonale de ces cellules résistantes au traitement est favorisée sous IM par rapport à celle des autres clones malins. C’est pourquoi, suivant le moment d’apparition de la mutation au cours du traitement, il est possible d’observer une rémission complète, une résistance précoce, ou une rechute après une réponse initiale.