Résumés
Résumé
La médecine classique ne sait pas réparer les tissus vitaux comme le foie, le cerveau, les muscles ou le pancréas lorsqu’ils sont atteints par un processus pathologique ou une dégénérescence liée à l’âge. Même si ces tissus contiennent des cellules souches, celles-ci ne remplissent pas cette mission de réparation spontanément, et on ne sait ni les isoler, ni les activer ex vivo. On comprend donc l’enthousiasme soulevé par l’observation selon laquelle les cellules de la moelle osseuse - qui contient entre autres les cellules souches hématopoïétiques utilisées en transplantation depuis des décennies - participaient à la formation d’autres tissus, dont le foie, le muscle et le cerveau, et bien d’autres encore. C’était le début de la fulgurante, mais éphémère, carrière de deux mots, « plasticité » et « transdifférenciation », mais également d’une campagne médiatique imprudente et source de grande confusion, promettant pour demain la régénération de tous nos organes, espoir déguisé d’immortalité. Maintenant que les mécanismes sous-jacents à ces observations commencent à être mieux compris, que la réalité d’une application thérapeutique efficace immédiate s’estompe, on peut tenter de faire, dans la sérénité, le point sur ces acteurs virtuels que sont les cellules souches, sur ce que nous disent réellement les expériences, et pourquoi elles ont entraîné tant de controverses.
Summary
Recent unexpected observations in adult rodents that stem/progenitor cells located in the bone marrow, but also in other tissues, could, after their transplantation to an irradiated host contribute to the regeneration of damaged organs such as brain, liver, pancreas or muscle, have raised much hope for future therapeutic applications. These data have also initially been interpreted as a proof of a possible transdifferentiation or plasticity of adult stem cells located in these tissues. Additional experiments rigorously analyzed have tempered initial enthusiasm, by showing that if marrow cells do migrate in damaged muscles and liver, their contribution to organ repair is low, and in some cases, explained by cell fusion. Nevertheless, among bone marrow cells, two categories of stem cells now emerge that have a potentially tremendous interest in cell therapy, if we succeed in understanding how to purify, amplify and differentiate these more efficiently and reproducibly.
Corps de l’article
Une histoire surprenante
En janvier 1999, une publication tonitruante de Bjornson dans Science prétendait que des cellules de « neurosphères » - agrégats cellulaires sphériques résultant de la prolifération in vitro de progéniteurs immatures issus du cerveau (striatum) de souris adultes - produisaient des cellules sanguines fonctionnelles lorsqu’elles étaient injectées par voie intraveineuse à une souris irradiée [1]. C’était le brain to blood, mettant au grand jour une possible transgression des dogmes selon lesquels une cellule souche nichée dans un tissu donné n’engendre que les seules cellules spécialisées de ce tissu, et ne peut pas adopter dans sa descendance le destin de deux feuillets embryonnaires différents. Depuis 15 ans, on sait que les cellules souches mésenchymateuses côtoient les cellules souches hématopoïétiques dans la moelle osseuse et, depuis 1997, on avait quelques soupçons sur la participation de cellules de la moelle osseuse à de nombreux tissus, cellules gliales dans le cerveau [2, 3], mais aussi cellules musculaires [4, 5] ((→) m/s 1998, n° 6-7, p. 803). Mais il semble que l’article de Science ait joué le rôle de catalyseur entre 1999 et 2002, inaugurant une série de descriptions inattendues: marrow to liver [6-8], marrow to muscle [9], muscle to blood [10, 11] et blood to brain [12, 13].
Le plus surprenant est de n’avoir fait ces observations que récemment, alors que le protocole expérimental utilisé est celui, fort simple, d’une transplantation, par voie intraveineuse, de cellules de la moelle osseuse (ou d’un autre tissu) d’un donneur mâle à un receveur femelle généralement irradié. Plusieurs semaines ou plusieurs mois après la greffe, les tissus du receveur sont analysés par immunohistochimie, à la recherche de cellules issues du donneur et co-exprimant des marqueurs spécifiques des tissus étudiés. La surprise a été de constater, après transplantation médullaire, la présence dans plusieurs organes comme le foie [7, 9, 14], le cerveau [12, 13] ou le muscle [4, 5, 8] ((→) m/s 1999, n° 12, p. 1427), et maintenant beaucoup d’autres, en particulier ceux ayant une composante épithéliale (poumon, intestin, rein…) [15-17] ((→) m/s 2002, n° 2, p. 164), de cellules spécialisées de ce tissu mais provenant du donneur, et donc d’un tissu, la moelle, qu’on ne soupçonnait pas être investie d’un tel potentiel. Aussi stupéfiant, l’établissement d’un chimérisme hématopoïétique a été montré après greffe de cellules provenant du muscle [10, 11] ou du cerveau [1]. Ces observations, faites initialement chez le rongeur, puis, pour certaines, confirmées chez l’homme (examen post-mortem de tissus de receveurs de greffes de moelle osseuse), démontraient donc que des cellules souches présentes dans un tissu donné, et dont on pensait le potentiel restreint à ce seul tissu, étaient capables d’en exprimer un autre, pour peu qu’elles soient exposées à un environnement différent. C’est ce que désignaient les termes « plasticité » et/ou « transdifférenciation ». Malgré des mises en garde précoces [18, 19] contre une interprétation imprudente et peu rigoureuse d’observations expérimentales très préliminaires et incomplètement explorées, leur exploitation médiatique a un temps investi l’homme du privilège des salamandres ((→) m/s 2002, n° 10, p. 917), c’est-à-dire d’une possible autorégénération facile de ses organes malades [20, 21].
Les analyses plus rigoureuses effectuées depuis, y compris par les auteurs des publications initiales, ont confirmé la solidité de « vieux concepts » qui ne s’en laissent pas conter, et n’ont trouvé aucune preuve de « transdifférenciation » stricto sensu, dont la réalité s’estompe. En revanche, clin d’oeil au « clonage thérapeutique », la formation d’hétérocaryons par fusion cellulaire, un événement fréquent chez l’adulte, offre un autre exemple de transfert nucléaire et de re-programmation aux effets bénéfiques, mais dont l’exploitation thérapeutique est également très incertaine. Heureusement, il nous reste à confirmer l’existence d’une nouvelle cellule souche et à tirer au clair d’autres observations dont la possibilité d’un potentiel hématopoïétique extra-médullaire. Ironie rassurante, tous ces travaux ne doivent rien à la technologie, mais tout aux approches « ancestrales » de biologie cellulaire où l’oeil, l’imagination et le temps sont irremplaçables.
Qu’est-ce qu’une cellule souche?
À l’exception des embryologistes et des scientifiques s’occupant de plantes ((→) m/s 2001, n° 8-9, p. 836) ou de « mouches » ((→) m/s 2001, n° 5, p. 628), ceux d’entre nous qui étudient les cellules souches adultes n’en ont jamais vu. Elles sont rares et n’expriment aucun marqueur de surface spécifique [22, 23]. Elles partagent certains antigènes (CD34, Thy-1, CD133, Flk-1, Sca-1, c-kit) qui ne leur sont pas spécifiques. Deux propriétés les distinguent de leur descendance plus mature: beaucoup sont capables d’exclure le colorant vital Hoechst (Ho) 33342, qui se fixe sur les molécules d’ADN et, de ce fait, permet d’analyser le cycle cellulaire. Mais si l’analyse de l’émission de fluorescence du Ho excité dans l’ultraviolet (350 nm) est faite simultanément dans deux longueurs d’onde (450 et 675 nm), on peut identifier une petite population de cellules très faiblement fluorescentes, appelées SP (side population) [24]. Ces cellules SP expulsent le colorant grâce à l’expression de transporteurs de la famille ABC (ATP-binding cassette transporter) ou MDR (multidrug resistance) et sont, dans plusieurs tissus, très enrichies en cellules souches. Enfin, la quiescence des cellules souches, identifiable par leur capacité de rétention d’intercalants de l’ADN comme le BrdU (administré à l’animal ou aux cultures), est souvent utilisée pour les reconnaître sur coupes histologiques (LRC, label retaining cells), notamment dans l’épiderme, l’intestin ou le cerveau, et les distinguer de leurs descendants immédiats, les transit-amplifying cells, ou progéniteurs en phase proliférative [25].
Dans certains tissus où les cellules souches sont profondément enfouies, leur localisation anatomique très précise et leur rétention de BrdU permettent de les repérer histologiquement et d’en suivre le devenir (Figure 1). Ainsi, les cellules souches communes aux kératinocytes et aux composants du poil se nichent au niveau de l’implantation du muscle érecteur du poil [26], les cellules souches intestinales presque au fond de la crypte intestinale [27], les cellules progénitrices du muscle dans la zone bordant la fibre musculaire, d’où leur nom de satellites [28], les cellules souches neurales dans la zone sous-ventriculaire du plancher du IVe ventricule et dans le gyrus denté de l’hippocampe ((→) m/s 2001, n° 10, p. 1089) [25], et les cellules ovales au niveau des canaux de Hering [29]. Mais il en va différemment pour les cellules souches hématopoïétiques et mésenchymateuses, accessibles à la purification, car la moelle osseuse est un tissu « fluide » et que les premières circulent en grand nombre [30] (Figure 1).
Figure 1
Localisation anatomique des cellules souches dans les différents tissus de l’organisme.
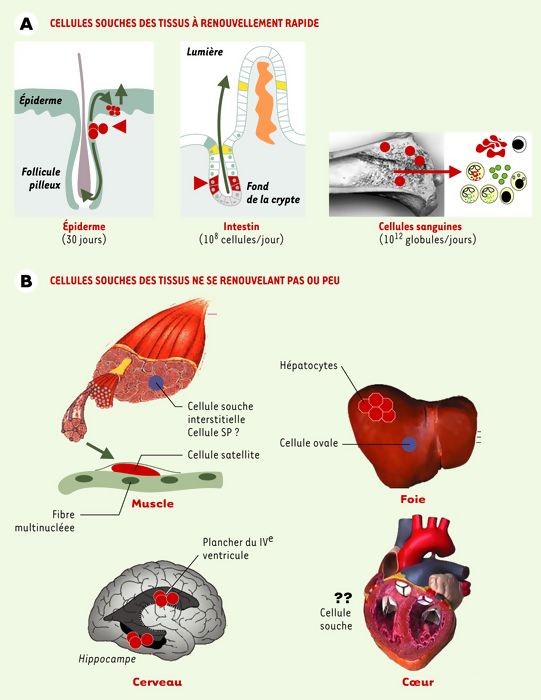
A. Trois tissus ont un renouvellement rapide (1-2 mois): les cellules souches y fonctionnent ainsi en permanence pour renouveler les cellules de la peau (et du système pileux), des villosités intestinales et du système hématopoïétique. Dans la peau et la crypte intestinale, les cellules souches ont été localisées de façon précise (pointes de flèche rouges) sur coupes histologiques, notamment grâce à leur capacité de rétention d’un intercalant de l’ADN (BrdU). Les flèches vertes indiquent les directions de la migration des cellules vers la surface épidermique et la lumière intestinale lors de leur différenciation. B. Dans les tissus quiescents, à faible renouvellement, des cellules souches sont présentes, leur localisation est également précise, mais leur fonction est moins bien définie. On en distingue deux types dans le muscle (les cellules satellites, peut-être distinctes des cellules interstitielles SP) ainsi que dans le foie (les cellules ovales et les hépatocytes, si l’on admet que les hépatocytes ont une fonction de cellule souche). Dans le cerveau, le marquage BrdU a localisé des cellules souches dans le plancher du IVe ventricule et dans le gyrus denté de l’hippocampe. Quant au coeur, on peut se poser la question de la signification d’une cellule souche.
Cette absence de critères de reconnaissance spécifiques et leur difficile accès tissulaire expliquent l’impossibilité de purifier à homogénéité les cellules souches, et donc les limites de l’analyse transcriptionnelle de ces populations. Aucun transcrit spécifique ne distingue cette fonction « souche », comme le montrent plusieurs analyses récentes du transcriptome de cellules enrichies en CSH (cellules souches hématopoïétiques), NSC (neural stem cells) ou ES (cellules souches embryonnaires), mais une communauté de gènes dont la transcription est particulièrement active à ce stade immature et qui contrôlent le métabolisme des ARN, la réparation de l’ADN, les voies de signalisation en aval du récepteur Notch, des récepteurs de cytokines, et les effecteurs du cycle cellulaire [31, 32]. Quel que soit le tissu considéré, faute de critères de reconnaissance spécifiques, les cellules souches ne peuvent donc être identifiées que par leur fonction [33], ce qu’écrivait déjà Chris Potten en 1990 [22] (voir encadré, page suivante). La fonction des cellules souches est de proliférer et de créer chez l’embryon, ou pérenniser chez l’adulte, la diversité des compartiments fonctionnels d’un tissu durant la vie de l’individu, en produisant un large spectre de cellules différenciées.
Bien avant d’en prouver l’existence expérimentalement, on avait intuitivement postulé l’existence de cellules souches multipotentes chez l’adulte pour expliquer le renouvellement permanent et rapide (environ tous les mois) d’au moins trois de nos tissus: la peau, l’intestin et le tissu hématopoïétique. La propriété d’autorenouvellement, qui colle historiquement au terme « cellule souche », mais reste le plus souvent invérifiable [34], avait l’avantage d’expliquer comment un si petit nombre de cellules pouvait fonctionner si longtemps (une seule cellule souche épidermique peut reconstituer une « unité » épidermique, moins de 4-5 cellules souches intestinales l’ensemble d’une villosité intestinale et une CSH l’ensemble des lignées sanguines). Aucune de ces deux propriétés - multipotence, et autorenouvellement - n’est absolument requise pour affirmer le caractère « souche » d’une cellule ; celle-ci doit en revanche fonctionner à long terme, et donc être capable d’un grand nombre de divisions cellulaires. C’est cette propriété qui distingue une cellule souche de ses descendants directs, les « progéniteurs », dont le nombre possible de divisions et le potentiel sont amputés en comparaison de ceux de la cellule parentale. Pourtant, l’usage a abusivement réuni ces deux catégories cellulaires sous la même appellation de « cellule souche ».
Toute la difficulté est donc d’ordre expérimental. Acquérir la preuve qu’une cellule est « souche » nécessite de caractériser sa descendance, in vitro et/ou in vivo: il s’agit donc d’une identification indirecte et rétrospective (Figure 2). Celle-ci doit répondre à deux exigences: l’une est de placer cette cellule souche dans des conditions qui permettent l’expression de toutes ses capacités de prolifération et de différenciation [33], ce qui n’est pas simple, compte tenu de la multiplicité des « environnements » cellulaires requis pour chaque voie de différenciation, de leur spécificité, et de leur fréquente incompatibilité. La seconde exigence est d’analyser les cellules individuellement, par la manipulation de cellules uniques ou le suivi d’un marqueur de clonalité [23, 35, 36]. Cette analyse clonale est imposée par l’hétérogénéité des tissus de départ et par l’impossibilité d’une purification à homogénéité des cellules souches, un phénotype n’étant jamais le reflet fidèle d’une fonction. Si ces contraintes expérimentales ne sont pas satisfaites, il est impossible de définir avec certitude le potentiel des cellules analysées, et donc à plus forte raison de conclure à leur « transdifférenciation ». Or, ces contraintes sont rarement satisfaites, ce qui devrait nous inciter à plus de prudence dans nos conclusions: comment être sûr que les conditions expérimentales choisies permettront à une CS d’exprimer l’ensemble de ses potentiels, puisqu’on ne les connaît pas a priori? Comment être sûr aussi qu’on n’altère pas ces propriétés par la manipulation des cellules ou des animaux receveurs?
Figure 2
Identification expérimentale des cellules souches.

L’identification d’une cellule souche ne peut se faire que de manière rétrospective et indirecte, par la mise en évidence de ses propriétés de prolifération et de différenciation. L’expression de ces propriétés in vitro ou in vivo (après transfert à l’animal) sera reconnue par la production de cellules différenciées identifiables par leur phénotype (cytométrie, histologie) ou par leur fonction. L’animal receveur a généralement un avantage sélectif qui facilite le développement des cellules greffées, irradiation pour détruire les populations endogènes et libérer les « niches » dans le cas des cellules satellites ou des cellules hématopoïétiques, ou lésion de l’organe cible. In vitro, la différenciation des cellules souches ne peut se faire qu’en présence de molécules appropriées, cytokines ou signaux émanant de cellules stromales, reproduisant l’équivalent de l’environnement qui prévaut in vivo.
Cellules souches vraies, cellules souches virtuelles?
Les cellules souches hématopoïétiques représentent peut-être le seul exemple de cellules souches multipotentes physiologiques identifiées de façon indiscutable à l’échelon clonal, in vivo comme in vitro chez l’animal adulte. Une seule cellule, de phénotype Sca-1+c-kit+Lin-Thy-1±, injectée à une souris irradiée, ou cultivée in vitro, peut produire l’ensemble des lymphocytes et des cellules myéloïdes différenciées nécessaires à la restauration définitive d’un statut hématologique normal, mais également des cellules souches capables à leur tour de reconstituer le système hématopoïétique d’un receveur secondaire irradié [23, 35]. Il ne s’agit pas stricto sensu d’un « autorenouvellement », puisqu’on ne peut pas démontrer l’identité moléculaire de la cellule fille et de la cellule parentale, mais d’un autorenouvellement « fonctionnel » ou plutôt d’une capacité de reconstitution « à long terme » [34]. Chez l’homme, la multipotence lymphoïde et myéloïde de cellules uniques a été démontrée in vitro, comme in vivo dans un modèle murin, sans toutefois que l’on puisse affirmer l’identité de ces cellules et de celles qui fonctionnent physiologiquement ou après greffe [36].
Toujours dans la moelle osseuse, une seconde population parfaitement identifiée est celle des cellules souches mésenchymateuses, à l’origine des cellules osseuses, cartilagineuses, stromales, adipocytaires et probablement musculaires (Figure 3); leur multipotence a été prouvée à l’échelon unicellulaire [37], et elles font déjà partie de l’arsenal thérapeutique des orthopédistes [38] ((→) m/s 2001, n° 1, p. 128). Une cellule aux propriétés similaires vient d’être décrite dans la membrane synoviale [39].
Figure 3
Filiation proposée pour les différentes populations de cellules présentes dans la moelle osseuse.

Ce schéma, qui fait des MAPC (multipotent adult progenitor cell) les ancêtres de toutes les populations hématopoïétiques et mésenchymateuses présentes dans la moelle osseuse, est encore spéculatif. En particulier, la filiation des progéniteurs endothéliaux et de l’hémangioblaste est encore mal définie. CS: cellule souche; GR: globule rouge; PN: polynucléaires neutrophiles; NK: natural killer; Mo: monocytes; Meg: mégacaryocytes; T, B: lymphocytes T et B; Dend: cellules dendritiques.
Une troisième population, les MAPC (multipotent adult progenitor cell), dont l’identification en juillet 2002 constitue la véritable percée de ces dernières années, ont un potentiel infiniment plus vaste que celui des deux précédentes [40], proche cette fois de celui de cellules ES, ce qui en fait une cible thérapeutique majeure (Figures 3 et 4). Leur localisation n’est probablement pas restreinte à la moelle osseuse [41]. In vivo, après leur injection à un receveur irradié, elles peuvent coloniser de multiples tissus et adopter leurs caractéristiques phénotypiques et fonctionnelles, confirmant qu’une cellule adulte peut adopter le destin de cellules issues pendant la vie embryonnaire de feuillets embryonnaires différents. Placée in vitro dans des conditions adéquates en cytokines et nutriments, ces MAPC adoptent un destin d’hépatocytes (endodermiques), de cellules endothéliales et hématopoïétiques (mésodermiques) et probablement de neurones et de cellules gliales (ectodermiques) (Figure 4). In vivo, micro-injectée dans un blastocyste de souris, une seule de ces cellules contribue à la formation de tous les tissus du nouveau-né, à l’exception du tissu nerveux. Surtout, placée dans un milieu pauvre en sérum additionné de cytokines comme le FGF (fibroblast growth factor), le PDGF (platelet-derived growth factor) et le LIF (leukemia inhibitory factor) et dans des conditions de densité cellulaire faible, une telle cellule peut proliférer à l’état indifférencié sans subir de sénescence réplicative pendant plus de 100 doublements (Figure 4). Ce comportement lui confère un intérêt thérapeutique majeur, parce qu’il permettrait d’accumuler un nombre important de cellules indifférenciées pouvant être conservées dans cet état, et dont on pourrait induire la différenciation en fonction du besoin. C’est l’expérimentateur qui, par son choix des conditions de culture, imposerait à la cellule son devenir. De telles caractéristiques les apparentent fonctionnellement aux cellules ES embryonnaires, avec lesquelles elles partagent certains marqueurs moléculaires restreints jusqu’à maintenant aux cellules souches ES (Oct-4, Rex), et donc absents des cellules souches adultes connues. L’étude a été réalisée à l’échelon clonal, ce qui garantit la réalité de la diversité des potentiels, mais il reste à reproduire les résultats, ce qui ne semble pas si simple, et éliminer tout autre artéfact [42-46].
Figure 4
Identification des MAPC (multipotent adult progenotor cell) chez la souris [35].
![Identification des MAPC (multipotent adult progenotor cell) chez la souris [35].](/fr/revues/ms/2003-v19-n6-7-ms537/006828ar/media/006828arf004n.jpg)
A. Les cellules médullaires n’exprimant ni l’antigène panleucocytaire CD45 (CD45-) ni la glycophorine A (TER119-) sont tout d’abord fractionnées, puis ensemencées à faible densité en présence d’un milieu pauvre en sérum de veau foetal (SVF) supplémenté avec les cytokines indiquées. B. Dans ces conditions, les cellules prolifèrent sans se différencier pendant plusieurs semaines et n’expriment pratiquement aucun marqueur. C. 1. La différenciation des cellules est induite par une modification du milieu de culture et par l’ajout des cytokines spécifiques des voies de différenciation choisies. 2. Les cellules peuvent également être injectées individuellement dans la masse interne d’un blastocyste réimplanté ensuite dans l’utérus d’une femelle gravide, ce qui permettra d’évaluer chez le nouveau-né leur contribution à la formation des différents tissus de l’organisme (la proportion de chimères obtenue est indiquée ainsi que l’importance du chimérisme). 3. Les cellules peuvent également être injectées par voie intraveineuse à un receveur immunodéficient (souris NOD-SCID, non obese diabetic-severe combined immunodeficient) et coloniser différents tissus du receveur. VEGF: vascular endothelial growth factor; FGF: fibroblast growth factor; BDNF: brain derived neurotrophic factor; HGF: hepatocyte growth factor; LIF: leukemia inhibitory factor; PDGF: platelet derived growth factor; EGF: epidermal growth factor; SVF: sérum de veau foetal.
Les propriétés des cellules souches identifiées dans d’autres tissus sont beaucoup plus floues, en partie parce que la structure de ces organes est un obstacle à leur dissociation facile (contrairement à la moelle osseuse), mais aussi parce que manquent, in vivo comme in vitro, les systèmes d’évaluation adéquats pour ces cellules. Comment définir une cellule souche dans le cerveau adulte, organe où le niveau de diversification fonctionnelle est tellement plus complexe que celui du tissu hématopoïétique ou hépatique? Peut-on se contenter de la production de trois types cellulaires phénotypiquement différents (neurones, astrocytes et oligodendrocytes) comme définition de la « multipotence »? Doit-on exiger la création d’une diversité fonctionnelle en principe terminée à la naissance? (voir la Nouvelle de M. Lemasson et P.M. Lledo, p. 664 de ce numéro). Les « neurosphères », expression de la prolifération clonale et de l’autorenouvellement de cellules mutipotentes, qui pour certains représentent des progéniteurs et non pas des cellules souches, constituent un « artéfact » de culture qui n’a aucune contrepartie in vivo [47]. La dérive de ces cellules, qui prolifèrent « à l’infini » en réponse à des cytokines puissantes comme le FGF ou l’EGF (epidermal growth factor), pourrait expliquer qu’elles aient pu acquérir un potentiel aberrant à l’origine des observations de Björnson en 1999, observation qui n’a pour l’instant jamais été reproduite [48].
Le foie constitue un cas à part: d’après les critères énoncés ci-dessus, il ne serait pas faux de qualifier les hépatocytes, dont on connaît la remarquable (et unique) capacité de réparation du tissu hépatique, de « cellules souches ». Quant aux cellules ovales (voir l’encadré de Y. Laperche, p. 697 de ce numéro), très rares, encore mystérieuses, elles constitueraient une sorte de réserve de « cellules souches/progéniteurs » prenant le relais si les hépatocytes ne pouvaient remplir leur mission de réparation [29]. La cellule souche pancréatique adulte reste quant à elle une énigme (voir l’encadré de R. Scharfmann, p. 695 de ce numéro). Enfin, dans le muscle, les cellules satellites sont indiscutablement capables, en cas de besoin, de donner naissance à des myoblastes qui fusionneront en myotubes, mais leur capacité de prolifération est limitée et leur potentiel restreint aux cellules musculaires (voir l’encadré de V. Mouly, p. ???). Dans les espaces situés entre les fibres musculaires, on identifie un autre progéniteur, de phénotype CD45-CD34+Sca-1+, peut-être proche des cellules SP excluant le Hoechst, et dont le potentiel dépasse la seule lignée musculaire et serait proche de celui des cellules mésenchymateuses [49]. Cellules ovales et satellites sont peut-être plus proches de progéniteurs déjà spécialisés que de cellules souches véritables [28]. Il est enfin intéressant de constater que les hépatocytes peuvent être tétraploïdes normalement et que les cellules différenciées du muscle, les myotubes, sont des cellules multinucléées, dont la genèse implique une étape de fusion cellulaire, un processus qui est à l’origine de beaucoup des données abusivement interprétées comme une « transdifférenciation » (voir plus loin).
Pour certains organes, comme le coeur ou le rein, dont la fonction impose, plus que la spécialisation d’un seul type cellulaire, l’organisation tridimensionnelle complexe de cellules épithéliales, endothéliales et musculaires, on peut s’interroger sur la notion de cellule souche. L’amélioration de la performance ventriculaire post-infarctus après administration de précurseurs endothéliaux et/ou musculaires (y compris squelettiques) qui n’ont pas de spécificité cardiaque en sont une illustration.
La moelle osseuse: une caverne d’Ali-Baba?
La moelle osseuse, comme tous les tissus, contient de multiples cellules souches et progéniteurs. L’expression « cellules souches médullaires » ne désigne pas une entité unique, et la méconnaissance de cette hétérogénéité a beaucoup contribué aux fausses interprétations qui ont pu être données des expériences récentes de transplantation médullaire.
Il y a dans la moelle osseuse (Figure 5) un compartiment extra vasculaire contenant des cellules souches très minoritaires ayant chacune un potentiel de différenciation spécifique et, pour certaines d’entre elles, leur descendance, progéniteurs (cellules dont le potentiel de prolifération et de différenciation est déjà restreint) et précurseurs (différenciés et reconnaissables morphologiquement). Mais la moelle est très vascularisée et, à ces populations présentes dans le parenchyme extravasculaire, il faut ajouter celles qui circulent en intravasculaire, mais ne résident pas dans la moelle osseuse [30]; elles contaminent les prélèvements de moelle osseuse (ou tout autre tissu, musculaire, par exemple). Peut-être est-ce le cas des cellules de type « ovales hépatiques » dont la présence vient d’être décrite dans la moelle osseuse [50]?
Figure 5
Anatomie de la moelle osseuse.
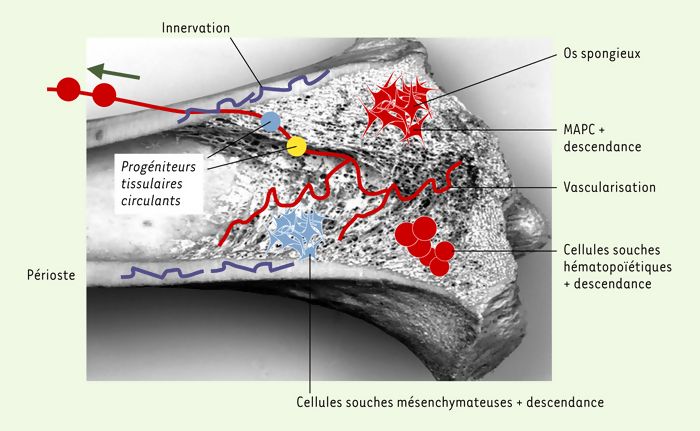
Le tissu médullaire présent dans la cavité médullaire des os longs est très vascularisé (vaisseaux représentés en rouge), et de nombreuses populations cellulaires, dont des cellules souches, y circulent en permanence et contaminent inévitablement tout prélèvement médullaire. Dans le parenchyme extravasculaire, on trouve au moins trois populations de cellules souches, les MAPC (multipotent adult progenitor cell), les cellules souches mésenchymateuses et les cellules souches hématopoiétiques. Ces cellules sont extrêmement minoritaires par rapport à leur descendance, représentée par les progéniteurs et les précurseurs.
C’est en ayant à l’esprit cette structure tissulaire que doivent être interprétées les observations, faites chez l’animal comme chez l’homme, démontrant l’origine médullaire de cellules ovales et d’hépatocytes, de cellules musculaires (satellites et myotubes), de neurones ou de cellules épithéliales contribuant à des tissus aussi divers que l’épithélium buccal, le rein ou les poumons. Quelle est, dans la moelle osseuse, l’origine de ces cellules? S’agit-il de progéniteurs hépatiques ou musculaires spécifiques résidant ou transitant dans la moelle osseuse, ou s’agit-il de l’expression du potentiel d’une cellule souche multipotente? Dans ce dernier cas, s’agit-il d’une CSH, d’une MAPC ou d’une cellule souche mésenchymateuse? Ou, comme cela est maintenant démontré, d’un hétérocaryon résultant de la fusion, in situ, d’une cellule hématopoïétique différenciée (probablement un macrophage tissulaire issu naturellement de la moelle osseuse) avec une cellule tissulaire différenciée?
L’implication des CSH classiques dans la production d’hépatocytes et de neurones est maintenant écartée, et elle l’est dans le cas du foie par le groupe de Weismann qui l’avait initialement suggérée. Deux arguments plaident en cette défaveur. Des cellules de phénotype Lin-Sca-1+Thy-1±c-kit+ provenant de souris transgéniques pour le gène GFP, et injectées individuellement par voie intraveineuse à des souris irradiées, reconstituent l’ensemble des lignées hématopoïétiques, mais ne participent qu’exceptionnellement à des tissus non hématopoïétiques [35]. Pourtant, cette même équipe avait décrit, dans un modèle murin de cytolyse hépatique grave (modèle de tyrosinémie par déficit en fumaryl acétoacétate hydrolase, FAH), la restauration de la fonction hépatique après transplantation d’un petit nombre de cellules médullaires ayant un phénotype de CSH, et la présence d’hépatocytes exprimant des marqueurs du donneur [7] ((→) m/s 2001, n° 8-9, p. 936). Il s’avère aujourd’hui que ces « hépatocytes » d’origine médullaire étaient des hétérocaryons créés par la fusion entre des probables cellules de Kupffer provenant des CSH greffées et des hépatocytes malades; le fait remarquable est que cette fusion induit une reprogrammation des noyaux de macrophages que traduit la synthèse de FAH par les hétérocaryons, et que ces hépatocytes polyploïdes restaurent la fonction hépatique de receveurs Fah-/- secondaires, voire tertiaires [45, 46] ((→) m/s 2001, n° 4, p. 491).
D’autres groupes ont également décrit la contribution de cellules d’origine médullaire à des tissus épithéliaux après transplantation: dans les expériences de Krause, également réalisées avec des cellules uniques, la restauration hématopoïétique est complète, et la descendance des cellules injectées colonise des tissus épithéliaux tels que le poumon, l’intestin ou la peau, et y expriment des marqueurs épithéliaux [15] ((→) m/s 2003, n° 6-7, p. 671). Mais les équipes de Krause et Weismann ne travaillaient pas avec la même population de cellules médullaires. La première utilisait des cellules sélectionnées sur leur capacité de homing, dont on ne connaissait pas le phénotype immunologique: elles provenaient en effet de la moelle osseuse de receveurs primaires ayant reçu 48 heures auparavant des cellules de moelle osseuse Lin-. Il est donc vraisemblable que les populations utilisées dans les deux études soient différentes fonctionnellement, et que celles de l’équipe de Krause soient proches des MAPC de l’équipe de Verfaillie, ces cellules ayant le potentiel le plus large après transplantation [40]. La contribution d’une fusion cellulaire à ces observations n’a pas été mesurée.
Nous serions donc en présence de trois populations rares de cellules souches, toutes issues de la moelle osseuse, toutes multipotentes, toutes identifiées à l’échelon clonal, peut-être apparentées, et dont aucune n’a le même potentiel! C’est l’illustration de la difficulté qu’ont les biologistes cellulaires à comparer les propriétés de populations sélectionnées sur des critères différents, analysées in vivo dans des modèles animaux différents, avec des critères d’évaluation différents (la controverse récente publiée dans Science en février 2003 à propos des artéfacts liés à l’utilisation des traceurs comme la GFP ou LacZ en est une illustration), et interprétés dans des laboratoires différents.
Différenciation épithéliale ou fusion cellulaire, le problème est posé ici aussi. Les cellules mésenchymateuses de moelle osseuse peuvent fusionner avec les cellules épithéliales de l’arbre respiratoire dans une proportion qui peut atteindre 25 % [44], ou avec des myoblastes, contribuant à la constitution de myotubes. Mais dans le cas du chimérisme de l’épithélium buccal observé après greffe de cellules de moelle osseuse, qui atteint parfois des proportions importantes (jusqu’à 12 % [16]), les auteurs ont pris grand soin d’éliminer la possibilité d’une fusion et l’affirment haut et fort. Une différenciation épithéliale de cellules d’origine médullaire est donc probable. Que celle-ci soit facilitée par des signaux émis en réponse à une lésion tissulaire est suggéré, et même parfois démontré. C’est ainsi le cas du muscle, dans lequel les cellules de moelle osseuse migrent dans les niches des cellules « satellites » et y restent quiescentes jusqu’à leur induction par une lésion musculaire [8]; ce rôle a, en revanche, été infirmé dans le cas du foie [51]. Plus que la lésion, c’est peut-être l’activité proliférative du tissu qui explique l’importance du chimérisme dans certains tissus, par exemple au niveau de l’épithélium buccal qui est en constant renouvellement [16]. Enfin, il ne faut pas négliger l’impact de l’irradiation du receveur, pratiquement constante, qui accroît la perméabilité vasculaire et facilite la pénétration tissulaire.
Les vieux concepts ont encore cours
À l’heure où ce texte est rédigé, nous n’avons pas une compréhension encore claire de tous les mécanismes mis en jeu. Mais il n’y aura probablement pas de révolution « conceptuelle » majeure. Nous sommes loin de l’axolotl et de l’hydre d’eau douce, deux petits organismes qui régénèrent facilement une partie sectionnée de leur corps [21, 52] en stimulant une réserve de cellules souches indifférenciées (hydre) [52] ou en déclenchant la dé-différenciation puis la re-différenciation de cellules différenciées (salamandre) [21]. Chez les rongeurs, modèle animal le plus souvent utilisé, et probablement chez l’homme, l’hétérogénéité tissulaire peut expliquer beaucoup des données exposées ci-dessus, et la situation est peut-être relativement simple: chaque tissu, dont la moelle osseuse, hébergerait trois types de cellules « souches ». En premier lieu, des cellules souches spécialisées qui ne peuvent emprunter qu’une voie de différenciation, celle qui aboutit aux cellules du tissu dans lequel elles résident (cellules SP et cellules satellites du muscle, cellules ovales du foie, CSH, cellules souches épidermiques, cellules souches sous-ventriculaires du système nerveux central, SNC). Certaines de ces cellules peuvent être vraiment qualifiées de « cellules souches » si elles engendrent une diversité fonctionnelle dans leur descendance (c’est le cas pour les CSH et les cellules neurales) et/ou peuvent fonctionner à long terme. Il existerait aussi dans de nombreux tissus des cellules souches multi-tissulaires, de type MAPC, dont on pourrait peut-être rapprocher les cellules souches mésenchymateuses, présentes également hors de la moelle osseuse et dont le potentiel est fort large [39]. Enfin, un prélèvement tissulaire peut être contaminé par des cellules souches circulantes présentes dans le compartiment vasculaire irriguant le tissu donné, et qu’il est souvent impossible d’éliminer ((→) m/s 2002, n° 3, p. 290).
Cette contamination explique peut-être la reconstitution hématopoïétique observée après transplantation de cellules du muscle squelettique. Les cellules satellites et autres progéniteurs musculaires sont très minoritaires dans un prélèvement de muscle, et il est possible, mais peut-être pas systématique, qu’une contamination par une CSH circulante, une CSH résidant dans le muscle (comme l’a démontré Ogawa [53, 54]) ou encore une cellule de type MAPC explique les résultats. Travailler avec des cellules individuelles est infiniment plus compliqué dans le cas du muscle que dans le cas de cellules médullaires [8].
Enfin, on sait que certaines cellules hématopoïétiques (notamment les macrophages) migrent dans tous les tissus, et plusieurs articles récents ont confirmé qu’un mécanisme de fusion cellulaire est bel et bien responsable d’une partie du chimérisme observé après greffe de moelle osseuse. Deux articles parus dans la revue Nature avaient semé le doute en 2000 en révélant la réalité du processus de fusion s’opérant entre cellules embryonnaires ES et cellules adultes en culture et conférant aux premières les propriétés des secondes [42, 43]. Ce mécanisme contribue effectivement au chimérisme observé après greffe de moelle osseuse dans des organes comme le cerveau [55, 56] ou le foie [45, 46]. Il semble que ce processus fusionnel suffise à lui seul à expliquer la restauration de la fonction hépatique des souris FAH-/- après greffe de moelle; ce ne serait pas le cas dans le cerveau, le muscle ou d’autres épithéliums, où un autre mécanisme interviendrait également; mais les difficultés techniques rencontrées dans l’évaluation de la ploïdie des noyaux incitent à la prudence.
Cellules souches: réalité thérapeutique?
Soulignons d’emblée qu’il importe de distinguer deux situations très différentes: la première est celle des maladies dégénératives disséminées, ou touchant un organe essentiel, que l’on pouvait espérer améliorer grâce à des cellules souches ayant le pouvoir intrinsèque de reconstituer l’ensemble des populations constituant l’organe. Une seconde situation, beaucoup plus réaliste, et d’ailleurs déjà exploitée, consiste à utiliser non pas des cellules souches, mais des progéniteurs ou des précurseurs déjà engagés dans une voie de différenciation et capables de réparer efficacement et rapidement un dégât limité (Figure 6).
Figure 6
Hiérachie des compartiments de cellules souches et de progéniteurs.

La hiérarchie des cellules figure dans la colonne de gauche: dans tous les tissus, les cellules souches multipotentes, douées d’une grande capacité de prolifération, donnent naissance à des progéniteurs, dont la prolifération et le potentiel sont déjà amputés, mais qui sont encore capables de remplacer efficacement des cellules spécialisées d’un tissu. Ces progéniteurs se divisent eux-mêmes en précurseurs, souvent morphologiquement reconnaissables, différenciés, mais peu capables de division et ayant une durée de vie très limitée. Chacune de ces trois populations peut être utilisée à des fins thérapeutiques: quelques exemples sont indiqués dans la colonne de droite.
Thérapies cellulaires utilisant des progéniteurs
Contrairement aux cellules souches, les progéniteurs sont fonctionnels en quelques divisions, mais ont une durée de vie limitée et ne peuvent donc théoriquement pas fonctionner à long terme et réparer un organe de façon définitive. Cela ne constitue pas un handicap dans le cas d’un tissu à renouvellement lent, ou dans une situation transitoire avant une réparation endogène, mais n’est pas applicable à la réparation d’un tissu à renouvellement rapide. Beaucoup de ces progéniteurs sont présents dans la moelle osseuse, accessible chez le patient, réalisant une situation de greffe « autologue » très favorable. C’est le cas des progéniteurs mésenchymateux, déjà utilisés par les orthopédistes, ainsi que des progéniteurs endothéliaux, dont l’origine est incertaine (hémangioblaste médullaire, dont l’existence même n’est pas prouvée expérimentalement, MAPC ou cellule souche mésenchymateuse), mais qui sont bien caractérisés phénotypiquement et fonctionnellement [57], et dont l’utilité dans le remodelage vasculaire des pathologies ischémiques [58] et de l’infarctus du myocarde [59, 60] a été récemment reconnue. On peut également en rapprocher deux démarches exceptionnelles, les greffes de neurones foetaux dans certaines maladies neurodégénératives comme la maladie de Hungtington [61], ou celles des îlots β du pancréas dans le diabète insulino-dépendant [62].
Utilisation de cellules souches
Si l’on exclut les CSH et les cellules souches épidermiques déjà très utilisées avec succès, il faut bien reconnaître que les données scientifiques récentes ne vont pas changer la donne à court terme.
Sur un plan théorique, on peut douter de l’efficacité d’une utilisation thérapeutique de cellules souches adultes issues de moelle osseuse (seul tissu concerné) dans un avenir proche: la contribution de cellules médullaires à la reconstitution d’un foie [51] ou d’un muscle lésé [63] est trop faible dans les expériences réalisées chez le rongeur pour être d’un quelconque bénéfice thérapeutique, et personne ne songe réellement à utiliser cette approche dans les maladies neurodégénératives. Que le chimérisme soit la conséquence d’une fusion cellulaire introduit une incertitude supplémentaire, et même si, expérimentalement, la « reprogrammation » ainsi créée dans les hétérocaryons restaure une fonction hépatique normale, leur instabilité génétique probable compromet leur avenir thérapeutique.
L’exploitation thérapeutique des propriétés des cellules de type MAPC reste en revanche très séduisante; il reste cependant nécessaire que d’autres équipes confirment l’immense potentiel de prolifération et de différenciation de ces cellules. Il faudrait alors apprécier le risque tumoral, dont on sait qu’il grève l’utilisation des cellules ES. Par ailleurs, cela nécessitera d’apprendre à maîtriser la migration des cellules greffées dans l’organe et leur activation locale [8], contrôlées par des signaux environnementaux probablement indépendants, mais dont nous ignorons à peu près tout et dont l’identification constitue une voie de recherche essentielle. On ne peut qu’insister sur cette démarche, d’autant qu’une utilisation thérapeutique de signaux activateurs de cellules souches ne se conçoit pas seulement dans le cadre d’une greffe de cellules isolées d’un autre organe, mais aussi dans celui de la stimulation de cellules souches endogènes.
Cette alternative ne doit en effet pas être négligée. On connaît depuis longtemps le pouvoir qu’ont les cellules souches hématopoïétiques endogènes de restaurer totalement le système hématopoïétique après sa destruction par une irradiation ou une chimiothérapie. On peut donc envisager une réaction identique pour d’autres tissus dans lesquels des cellules souches endogènes ont été identifées, en particulier dans le SNC ou le muscle. S’il est irréaliste de penser aujourd’hui à les purifier et à les amplifier ex vivo, il ne l’est probablement pas de les activer localement [64, 65]. Les succès récents dans les cas d’ischémie cérébrale ((→) m/s 2003, n° 3, p. 265) ou d’encéphalite auto-immune ((→) m/s 2003, n° 3, p. 263) sont prometteurs, et devraient stimuler les efforts visant à identifier les signaux de l’environnement responsables de l’activation des cellules souches et de leur migration intratissulaire sur le lieu du déficit.
Parties annexes
Remerciements
Merci à tous ceux et celles que j’ai côtoyés et qui, par leur discussion, enrichissent ma réflexion sur les cellules souches depuis si longtemps et à l’Inserm, l’ARC, l’AFM pour leur soutien à nos travaux.
Références
- 1. Bjornson CR, Rietze RL, Reynolds BA, Magli MC, Vescovi AL. Turning brain into blood: a hematopoietic fate adopted by adult neural stem cells in vivo. Science 1999; 283: 534-7.
- 2. Eglitis MA, Mezey E. Hematopoietic cells differentiate into both microglia and macroglia in the brains of adult mice. Proc Natl Acad Sci USA 1997; 94: 4080-5.
- 3. Azizi SA, Stokes D, Augelli BJ, DiGirolamo C, Prockop DJ. Engraftment and migration of human bone marrow stromal cells implanted in the brains of albino rats-similarities to astrocyte grafts. Proc Natl Acad Sci USA 1998; 95: 3908-13.
- 4. Ferrari G, Cusella-De Angelis G, Coletta M, et al. Muscle regeneration by bone marrow-derived myogenic progenitors. Science 1998; 279: 1528-30.
- 5. Gussoni E, Soneoka Y, Strickland CD, et al. Dystrophin expression in the mdx mouse restored by stem cell transplantation. Nature 1999; 401: 390-4.
- 6. Petersen BE, Bowen WC, Patrene KD, et al. Bone marrow as a potential source of hepatic oval cells. Science 1999; 284: 1168-70.
- 7. Lagasse E, Connors H, Al-Dhalimy M, et al. Purified hematopoietic stem cells can differentiate into hepatocytes in vivo. Nat Med 2000; 6: 1229-34.
- 8. Labarge MA, Blau HM. Biological progression from adult bone marrow to mononucleate muscle stem cell to multinucleate muscle fiber in response to injury. Cell 2002; 111: 589-601.
- 9. Theise ND, Badve S, Saxena R, et al. Derivation of hepatocytes from bone marrow cells in mice after radiation-induced myeloablation. Hepatology 2000; 31: 235-40.
- 10. Jackson KA, Mi T, Goodell MA. Hematopoietic potential of stem cells isolated from murine skeletal muscle. Proc Natl Acad Sci USA 1999; 96: 14482-6.
- 11. Seale P, Sabourin LA, Girgis-Gabardo A, et al. Pax7 is required for the specification of myogenic satellite cells. Cell 2000; 102: 777-86
- 12. Mezey E, Chandross KJ, Harta G, Maki RA, McKercher SR. Turning blood into brain: cells bearing neuronal antigens generated in vivo from bone marrow. Science 2000; 290: 1779-82
- 13. Brazelton TR, Rossi FM, Keshet GI, Blau HM. From marrow to brain: expression of neuronal phenotypes in adult mice. Science 2000; 290: 1775-9.
- 14. Korbling M, Katz R, Khanna A, et al. Hepatocytes and epithelial cells of donor origin in recipients of peripheral-blood stem cells. N Engl J Med 2002; 346: 738-46.
- 15. Stamm C, Westphal B, Kleine HD, et al. Autologous bone-marrow stem-cell transplantation for myocardial regeneration. Lancet 2003; 361: 45-6.
- 16. Tran SD, Pillemer SR, Dutra A, et al. Differentiation of human bone marrow-derived cells into buccal epithelial cells in vivo: a molecular analytical study. Lancet 2003; 361: 1084-8.
- 17. Ito T. Stem cells of the adult kidney: where are you from? Nephrol Dial Transplant 2003; 18: 641-4.
- 18. Morrison SJ. Stem cell potential: can anything make anything? Curr Biol 2001; 11: R7-9.
- 19. Anderson DJ, Gage FH, Weissman IL. Can stem cells cross lineage boundaries? Nat Med 2001; 7: 393-5.
- 20. Pearson H. The regeneration gap. Nature 2001; 414: 388-90.
- 21. Echeverri K, Tanaka EM. Ectoderm to mesoderm lineage switching during axolotl tail regeneration. Science 2002; 298: 1993-6.
- 22. Potten CS, Loeffler M. Stem cells: attributes, cycles, spirals, pitfalls and uncertainties. Lessons for and from the crypt. Development 1990; 110: 1001-2.
- 23. Osawa M, Hanada K, Hamada H, Nakauchi H. Hematopoietic stem cells. Science 1996; 273: 242-5.
- 24. Goodell MA, Brose K, Paradis G, Conner AS, Mulligan RC. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J Exp Med 1996; 183: 1797-806.
- 25. Morrison SJ, White PM, Zock C, Anderson DJ. Prospective identification, isolation by flow cytometry, and in vivo self-renewal of multipotent mammalian neural crest stem cells. Cell 1999; 96: 737-49.
- 26. Oshima H, Rochat A, Kedzia C, Kobayashi K, Barrandon Y. Morphogenesis and renewal of hair follicles from adult multipotent stem cells. Cell 2001; 104: 233-45.
- 27. Marshman E, Booth C, Potten CS. The intestinal epithelial stem cell. Bioessays 2002; 24: 91-8.
- 28. Cossu G, Mavilio F. Myogenic stem cells for the therapy of primary myopathies: wishful thinking or therapeutic perspective? J Clin Invest 2000; 105: 1669-74.
- 29. S. Sell. Heterogeneity and plasticity of hepatocyte lineage cells. Hepatology 2001; 33: 738-50.
- 30. Wright D, Wagers A, Gulati A, Johnson F, Weissman I. Physiological migration of hematopoietic stem and progenitor cells. Science 2001; 294: 1933-6.
- 31. Ivanova NB, Dimos JT, Schaniel C, Hackney JA, Moore KA, Lemischka IR. A stem cell molecular signature. Science 2002; 298: 601-4
- 32. Ramalho-Santos M, Yoon S, Matsuzaki Y, Mulligan RC, Melton DA. «Stemness»: transcriptional profiling of embryonic and adult stem cells. Science 2002; 298: 597-600
- 33. Morrison SJ, Shah NM, Anderson DJ. Regulatory mechanisms in stem cell biology. Cell 1997; 88: 287-98.
- 34. Lansdorp PM. Self-renewal of stem cells. Biol Blood Marrow Transplant 1997; 3: 171-8.
- 35. Wagers AJ, Sherwood RI, Christensen JL, Weissman IL. Little evidence for developmental plasticity of adult hematopoietic stem cells. Science 2002; 297: 2256-9.
- 36. Robin C, Pflumio F, Vainchenker W, Coulombel L. Identification of lymphomyeloid primitive progenitor cells in fresh human cord blood and in the marrow of nonobese diabetic-severe combined immunodeficient (NOD-SCID) mice transplanted with human CD34+ cord blood cells. J Exp Med 1999; 189: 1601-10
- 37. Pittenger M, Mackay A, Beck S, et al. Multilineage potential of adult human mesenchymal stem cells. Science 1999; 284: 143-7.
- 38. Noel D, Djouad F, Jorgense C. Regenerative medicine through mesenchymal stem cells for bone and cartilage repair. Curr Opin Investig Drugs 2002; 3: 1000-4.
- 39. De Bari C, Dell’Accio F, Vandenabeele F, Vermeesch JR, Raymackers JM, Luyten FP. Skeletal muscle repair by adult human mesenchymal stem cells from synovial membrane. J Cell Biol 2003; 160: 909-18.
- 40. Jiang Y, Jahagirdar BN, Reinhardt RL, et al. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature 2002; 418: 41-9.
- 41. Jiang Y, Vaessen B, Lenvik T, Blackstad M, Reyes M, Verfaillie CM. Multipotent progenitor cells can be isolated from postnatal murine bone marrow, muscle, and brain. Exp Hematol 2002; 30: 896-904.
- 42. Terada N, Hamazaki T, Oka M, et al. Bone marrow cells adopt the phenotype of other cells by spontaneous cell fusion. Nature 2002; 416: 542-5.
- 43. Ying Q, Nichols J, Evans E, Smith A. Changing potency by spontaneous fusion. Nature 2002; 416: 545-8.
- 44. Spees JL, Olson SD, Ylostalo J, et al. Differentiation, cell fusion, and nuclear fusion during ex vivo repair of epithelium by human adult stem cells from bone marrow stroma. Proc Natl Acad Sci USA 2003; 100: 2397-402.
- 45. Vassilopoulos G, Wang PR, Russell DW. Transplanted bone marrow regenerates liver by cell fusion. Nature 2003; 422: 901-4.
- 46. Wang X, Willenbring H, Akkari Y, et al. Cell fusion is the principal source of bone-marrow-derived hepatocytes. Nature 2003; 422: 897-901.
- 47. Doetsch F, Petreanu L, Caille I, Garcia-Verdugo Jm, Alvarez-Buylla A. EGF converts transit-amplifying neurogenic precursors in the adult brain into multipotent stem cells. Neuron 2002; 36: 1021-34.
- 48. Morshead CM, Benveniste P, Iscove NN, Van Der Kooy D. Hematopoietic competence is a rare property of neural stem cells that may depend on genetic and epigenetic alterations. Nat Med 2002; 8: 268-73.
- 49. Tamaki T, Akatsuka A, Anado K, et al. Identification of myogenic-endothelial progenitor cells in the interstitial spaces of skeletal muscle. J Cell Biol 2002; 157: 571-7.
- 50. Avital I, Inderbitzin D, Aoki T, et al. Isolation, characterization, and transplantation of bone marrow-derived hepatocyte stem cells. Biochem Biophys Res Commun 2002; 288: 156-64.
- 51. Wang E, Montini E, Al-Dhalimy M, Lagasse E, Finegold M, Grompe M. Kinetics of liver repopulation after bone marrow transplantation. Am J Pathol 2002; 161: 565-74.
- 52. Muller WA. Pattern formation in the immortal Hydra. Trends Genet 1996; 12: 91-6.
- 53. Kawada H, Ogawa M. Bone marrow origin of hematopoietic progenitors and stem cells in murine muscle. Blood 2001; 98: 2008-13.
- 54. Mckinney-Freeman S, Jackson K, Camargo F, et al. Muscle-derived hematopoietic stem cells are hematopoietic in origin. Proc Natl Acad Sci USA 2002; 99: 1341-6.
- 55. Mezey E, Key S, Vogelsang G, Szalayova I, Lange GD, Crain B. Transplanted bone marrow generates new neurons in human brains. Proc Natl Acad Sci USA 2003; 100: 1364-9.
- 56. Weimann JM, Charlton CA, Brazelton TR, Hackman RC, Blau HM. Contribution of transplanted bone marrow cells to Purkinje neurons in human adult brains. Proc Natl Acad Sci USA 2003; 100: 2088-93.
- 57. Reyes M, Dudek A, Jahagirdar B, et al. Origin of endothelial progenitors in human postnatal bone marrow. J Clin Invest 2002; 109: 337-46.
- 58. Orlic D, Hill JM, Arai AE. Stem cells for myocardal regeneration. Circ Res 2002; 91: 1092-102.
- 59. Stamm C, Westphal B, Kleine HD, et al. Autologous bone marrow stem cell transplantation for myocardial regeneration. Lancet 2003; 361: 45-6.
- 60. Orlic D, Kajstura J, Chimenti S, et al. Bone marrow cells regenerate infarcted myocardium. Nature 2001; 410: 701-5.
- 61. Bachoud-Levi AC, Remy P, Nguyen JP, et al. Motor and cognitive improvements in patients with Huntington’s disease after neural transplantation. Lancet 2000; 356: 1975-9.
- 62. Ryan EA, Lakey JR, Paty BW, et al. Successful islet transplantation: continued insulin reserve provides long-term glycemic control. Diabetes 2002; 51: 2148-57.
- 63. Ferrari G, Stornaiuolo A, Mavilio F. Failure to correct murine muscular dystrophy. Nature 2001; 411: 1014-5.
- 64. Nakatomi H, Kuriu T, Okabe S, et al. Regeneration of hippocampal pyramidal neurons after ischemic brain injury by recruitment of endogenous neural progenitors. Cell 2002; 110: 429-41.
- 65. Kruger GM, Morrison SJ. Brain repair by endogenous progenitors. Cell 2002; 110: 399-402
Liste des figures
Figure 1
Localisation anatomique des cellules souches dans les différents tissus de l’organisme.
A. Trois tissus ont un renouvellement rapide (1-2 mois): les cellules souches y fonctionnent ainsi en permanence pour renouveler les cellules de la peau (et du système pileux), des villosités intestinales et du système hématopoïétique. Dans la peau et la crypte intestinale, les cellules souches ont été localisées de façon précise (pointes de flèche rouges) sur coupes histologiques, notamment grâce à leur capacité de rétention d’un intercalant de l’ADN (BrdU). Les flèches vertes indiquent les directions de la migration des cellules vers la surface épidermique et la lumière intestinale lors de leur différenciation. B. Dans les tissus quiescents, à faible renouvellement, des cellules souches sont présentes, leur localisation est également précise, mais leur fonction est moins bien définie. On en distingue deux types dans le muscle (les cellules satellites, peut-être distinctes des cellules interstitielles SP) ainsi que dans le foie (les cellules ovales et les hépatocytes, si l’on admet que les hépatocytes ont une fonction de cellule souche). Dans le cerveau, le marquage BrdU a localisé des cellules souches dans le plancher du IVe ventricule et dans le gyrus denté de l’hippocampe. Quant au coeur, on peut se poser la question de la signification d’une cellule souche.
Figure 2
Identification expérimentale des cellules souches.
L’identification d’une cellule souche ne peut se faire que de manière rétrospective et indirecte, par la mise en évidence de ses propriétés de prolifération et de différenciation. L’expression de ces propriétés in vitro ou in vivo (après transfert à l’animal) sera reconnue par la production de cellules différenciées identifiables par leur phénotype (cytométrie, histologie) ou par leur fonction. L’animal receveur a généralement un avantage sélectif qui facilite le développement des cellules greffées, irradiation pour détruire les populations endogènes et libérer les « niches » dans le cas des cellules satellites ou des cellules hématopoïétiques, ou lésion de l’organe cible. In vitro, la différenciation des cellules souches ne peut se faire qu’en présence de molécules appropriées, cytokines ou signaux émanant de cellules stromales, reproduisant l’équivalent de l’environnement qui prévaut in vivo.
Figure 3
Filiation proposée pour les différentes populations de cellules présentes dans la moelle osseuse.
Ce schéma, qui fait des MAPC (multipotent adult progenitor cell) les ancêtres de toutes les populations hématopoïétiques et mésenchymateuses présentes dans la moelle osseuse, est encore spéculatif. En particulier, la filiation des progéniteurs endothéliaux et de l’hémangioblaste est encore mal définie. CS: cellule souche; GR: globule rouge; PN: polynucléaires neutrophiles; NK: natural killer; Mo: monocytes; Meg: mégacaryocytes; T, B: lymphocytes T et B; Dend: cellules dendritiques.
Figure 4
Identification des MAPC (multipotent adult progenotor cell) chez la souris [35].
A. Les cellules médullaires n’exprimant ni l’antigène panleucocytaire CD45 (CD45-) ni la glycophorine A (TER119-) sont tout d’abord fractionnées, puis ensemencées à faible densité en présence d’un milieu pauvre en sérum de veau foetal (SVF) supplémenté avec les cytokines indiquées. B. Dans ces conditions, les cellules prolifèrent sans se différencier pendant plusieurs semaines et n’expriment pratiquement aucun marqueur. C. 1. La différenciation des cellules est induite par une modification du milieu de culture et par l’ajout des cytokines spécifiques des voies de différenciation choisies. 2. Les cellules peuvent également être injectées individuellement dans la masse interne d’un blastocyste réimplanté ensuite dans l’utérus d’une femelle gravide, ce qui permettra d’évaluer chez le nouveau-né leur contribution à la formation des différents tissus de l’organisme (la proportion de chimères obtenue est indiquée ainsi que l’importance du chimérisme). 3. Les cellules peuvent également être injectées par voie intraveineuse à un receveur immunodéficient (souris NOD-SCID, non obese diabetic-severe combined immunodeficient) et coloniser différents tissus du receveur. VEGF: vascular endothelial growth factor; FGF: fibroblast growth factor; BDNF: brain derived neurotrophic factor; HGF: hepatocyte growth factor; LIF: leukemia inhibitory factor; PDGF: platelet derived growth factor; EGF: epidermal growth factor; SVF: sérum de veau foetal.
Figure 5
Anatomie de la moelle osseuse.
Le tissu médullaire présent dans la cavité médullaire des os longs est très vascularisé (vaisseaux représentés en rouge), et de nombreuses populations cellulaires, dont des cellules souches, y circulent en permanence et contaminent inévitablement tout prélèvement médullaire. Dans le parenchyme extravasculaire, on trouve au moins trois populations de cellules souches, les MAPC (multipotent adult progenitor cell), les cellules souches mésenchymateuses et les cellules souches hématopoiétiques. Ces cellules sont extrêmement minoritaires par rapport à leur descendance, représentée par les progéniteurs et les précurseurs.
Figure 6
Hiérachie des compartiments de cellules souches et de progéniteurs.
La hiérarchie des cellules figure dans la colonne de gauche: dans tous les tissus, les cellules souches multipotentes, douées d’une grande capacité de prolifération, donnent naissance à des progéniteurs, dont la prolifération et le potentiel sont déjà amputés, mais qui sont encore capables de remplacer efficacement des cellules spécialisées d’un tissu. Ces progéniteurs se divisent eux-mêmes en précurseurs, souvent morphologiquement reconnaissables, différenciés, mais peu capables de division et ayant une durée de vie très limitée. Chacune de ces trois populations peut être utilisée à des fins thérapeutiques: quelques exemples sont indiqués dans la colonne de droite.