Corps de l’article
La neuropathie sensitivomotrice héréditaire avec agénésie du corps calleux (NSMH/ACC) (OMIM 218000), également connue sous le nom d’agénésie du corps calleux avec polyneuropathie (ACCPN) ou syndrome d’Andermann, est une maladie à transmission autosomique récessive de début précoce. Les patients atteints de NSMH/ACC proviennent principalement de deux régions du Nord-Est du Québec, le Saguenay-Lac-Saint-Jean (SLSJ) et le comté de Charlevoix [1]. Un effet fondateur explique cette prédominance régionale. En reconstruisant la généalogie des familles atteintes, on arrive à la conclusion que des colons venus de France au XVIIe siècle (régions du Poitou et du Perche) sont à l’origine de cet effet fondateur [1], même si aucun cas n’a été à ce jour rapporté en France. Le gène responsable de cette maladie, SLC12A6, a été identifié récemment par notre laboratoire [2].
Cliniquement, NSMH/ACC se caractérise par un retard de développement moteur, une atteinte cognitive progressive, un dysmorphisme (palais ogival, syndactylie), une hypotonie, une amyotrophie, une aréflexie tendineuse, une polyneuropathie sensitivo-motrice, et un degré variable d’agénésie du corps calleux [3]. D’un point de vue évolutif, la maladie se caractérise par un retard du début de la marche (3,8 ans), une perte de l’autonomie ambulatoire vers 14 ans, et une scoliose précoce débutant à 10,4 ans. L’âge moyen de décès se situe vers 33 ans. Chez ces patients, les fonctions cérébrales sont souvent atteintes, et 39 % développent des épisodes psychotiques vers l’âge de 15 ans [3]. Les études électrophysiologiques montrent une certaine variabilité des vitesses de conduction motrices (16-57 m/s). Les conductions sensitives sont toujours abolies, même en bas âge. L’imagerie cérébrale est normale chez environ le tiers des patients, alors que les deux tiers présentent une agénésie partielle ou complète du corps calleux (Figure 1) [3]. On note à l’examen anatomopathologique - outre l’agénésie du corps calleux - une petite quantité de gonflements axonaux dans la substance blanche cérébrale et une préservation des faisceaux de Probst (faisceau longitudinal aberrant situé sur la face interne de l’hémisphère cérébral) (Figure 2) [4]. Il ne semble y avoir aucun signe ni d’atrophie corticale ni d’atteinte de la corne antérieure de la moelle épinière. On retrouve des axones gonflés dans les 3e et 7e nerfs crâniens, ainsi que dans les racines ventrales et postérieures de la moelle épinière. La biopsie du nerf sural montre une perte axonale et des axones gonflés.
Figure 1
Imagerie par résonance magnétique en coupe sagittale T1 montrant une agénésie complète du corps calleux chez un patient québécois de 23 ans atteint de neuropathie sensitivomotrice héréditaire avec agénésie du corps calleux (NSMH/ACC).
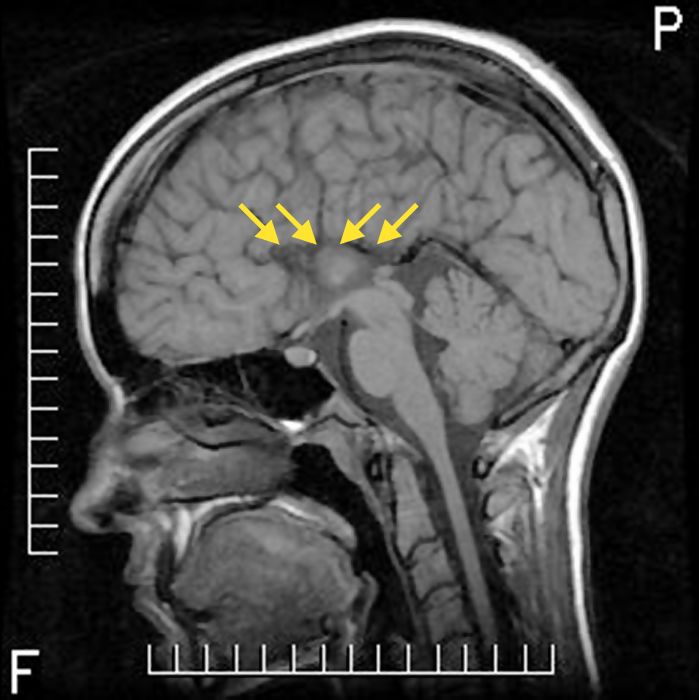
Une mutation (2436delG) de l’exon 18 du gène SLC12a6 a été identifiée chez ce patient. Les flèches jaunes indique l’emplacement habituel du corps calleux.
Figure 2
Coupe anatomopathologique coronale montrant une agénésie complète du corps calleux et une préservation du faisceau de Probst (flèche jaune) attaché au fornix.

Ce patient est décédé à 26 ans. Une mutation (2436delG) de l’exon 18 du gène SLC12a6 a été identifiée chez lui (cliché aimablement fourni par le Docteur Sterling Carpenter).
L’effet fondateur présent dans la NSMH/ACC au Québec a été confirmé sur le plan moléculaire. Nous avons ainsi démontré que 64 % des patients québécois atteints partageaient le même haplotype au sein d’une région du chromosome 15 s’étendant sur 4 cM, et que 97 % d’entre eux partageaient un même marqueur microsatellite (D15S1232) [1]. Une étude des gènes candidats présents dans la région a démontré que tous les patients québécois atteints présentaient une mutation ponctuelle de l’exon 18 (2436delG, Thr813fsX813) du gène SLC12A6 codant pour le canal potassique KCC3 [2]. Un seul patient québécois est porteur de la mutation mentionnée ci-dessus sur un allèle de l’exon 18 et d’une mutation de l’exon 11 sur un autre allèle (1584-1585delCTinsG, F529fsX531) [2]. Dix patients provenant de quatre autres pays (Italie, Autriche, Tanzanie, Turquie) présentaient des caractéristiques cliniques très semblables à celles des patients québécois. Une mutation ponctuelle du gène SLC12A6, située dans des exons différents de ceux atteints chez les patients québécois, a été confirmée chez deux de ces patients (Italie, exon 15; Turquie, exon 22) [2].
À l’heure actuelle, le rôle de la protéine KCC3 dans la physiopathologie de la NSMH/ACC n’est pas bien défini. De façon générale, KCC3 est un canal ionique qui agit comme co-transporteur K+-Cl-. Il est exprimé dans nombre de tissus (rein, muscle, système nerveux) [5]. La fonction cellulaire du KCC3 est controversée, certaines données suggérant qu’il intervient au niveau de la prolifération et de la mort cellulaires, d’autres au niveau de la régulation des concentrations ioniques (équilibre des ions Cl-). Lorsque la mutation présente dans la population québécoise (2436delG, Thr813fsX813) a été étudiée dans des ovocytes de Xenopus laevis, la protéine mutante était glycosylée et exprimée à la surface membranaire, mais elle n’était pas fonctionnelle [2]. Par ailleurs, un modèle de souris transgénique a été produit en remplaçant l’exon 3 du gène SLC12a6 par une cassette galactosidase/néomycine [2]. L’expression du KCC3 était nulle chez les souris homozygotes. Dès l’âge de deux semaines, ces souris présentaient un dysfonctionnement locomoteur marqué avec incoordination et faiblesse des membres, et l’analyse des fonctions cognitives supérieures montrait une diminution des fonctions liées à l’exploration de l’environnement. Par ailleurs, des études anatomiques détaillées du cerveau et de la moelle épinière n’ont pas révélé d’anomalies. Au niveau des nerfs sciatiques, on observait un grand nombre dgonflés, recouverts d’une fine couche de myéline. Ces anomalies des nerfs périphériques observées chez les souris transgéniques rappellent celles qui sont observées chez les patients atteints de la NSMH/ACC.
Au stade actuel de nos connaissances, nous estimons que KCC3 est impliqué tant au niveau du développement que du maintien du système nerveux central (SNC) et du système nerveux périphérique. Partant de l’agénésie du corps calleux [4], nous émettons l’hypothèse d’une action de KCC3 sur la migration axonale au cours du développement du système nerveux, puisque le corps calleux se forme entre la 11e et la 20e semaine de gestation [6]. De plus, la préservation du faisceau de Probst signifie que, dans l’agénésie qui caractérise la NSMH/ACC, les axones se forment mais sont incapables de traverser la ligne médiane étant donné l’absence de masse commissurale [6]. À ce stade, les études de localisation de la protéine dans le système nerveux ne nous permettent pas de conclure avec certitude si KCC3 agit principalement au niveau de l’axone ou de la myéline. La maladie suggère cependant une atteinte axonale, en raison de la présence des gonflements axonaux que l’on retrouve en général dans les maladies de l’axone [4]. Ces gonflements axonaux pourraient être secondaires à un dérèglement de l’équilibre ionique intracellulaire. Les dystrophies neuro-axonales et les axonopathies à cellules géantes sont aussi caractérisées par la présence de gonflements axonaux dans les nerfs périphériques [7], mais l’accumulation d’organites intracellulaires atypiques permet de les distinguer de la NSMH/ACC. Enfin, la NSMH/ACC se démarque facilement des autres NSMH par l’atteinte du système nerveux central.
La découverte du gène permettra une compréhension plus approfondie des mécanismes physiopathologiques de cette maladie, et peut-être aussi du développement et du fonctionnement du système nerveux, et pourrait contribuer à l’amélioration de l’approche thérapeutique. De plus, nous pourrons identifier d’autres cas à travers le monde afin de réaliser des études de corrélation phénotype-génotype. Aucun cas n’a été à ce jour rapporté en France, ce qui est d’autant plus surprenant que les études généalogiques faites au Québec montrent clairement que les familles porteuses de ces mutations ont des ancêtres communs venus de ce pays.
Parties annexes
Références
- 1. De Braekeleer M, Dallaire A, Mathieu J. Genetic epidemiology of sensorimotor polyneuropathy with or without agenesis of the corpus callosum in northeastern Quebec. Hum Genet 1993; 91: 223-7.
- 2. Howard H, Mount DB, Rochefort D, et al. The K-Cl cotransporter KCC3 is mutant in a severe peripheral neuropathy associated with agenesis of the corpus callosum. Nat Genet 2002; 32: 384-92.
- 3. Mathieu J, Bédard F, Prévost C, Langevin P. Neuropathie sensitivo-motrice héréditaire avec ou sans agénésie du corps calleux: étude radiologique et clinique de 64 cas. Can J Neurol Sci 1990; 17: 103-8.
- 4. Carpenter S. The pathology of the Andermann syndrome. In: Lassonde M, Jeeves M, eds. Callosal agenesis: a natural split brain? New York: Plenum Press, 1994 : 27-30.
- 5. Hiki K, D’Andrea R, Furze J, et al. Cloning, characterization, and chromosomal location of a novel human K-Cl cotransporter. J Biol Chem 1999; 274: 10661-7.
- 6. Dobyns W. Absence makes the search grow longer. Am JHum Genet 1996; 58: 7-16.
- 7. Mahadevan A, Santosh V, Gayatri N, et al. Infantile neuroaxonal dystrophy and giant axonal neuropathy: overlap diseases of neuronal cytoskeletal elements in childhood? Clin Neuropathol 2000; 19: 221-9.
Liste des figures
Figure 1
Imagerie par résonance magnétique en coupe sagittale T1 montrant une agénésie complète du corps calleux chez un patient québécois de 23 ans atteint de neuropathie sensitivomotrice héréditaire avec agénésie du corps calleux (NSMH/ACC).
Une mutation (2436delG) de l’exon 18 du gène SLC12a6 a été identifiée chez ce patient. Les flèches jaunes indique l’emplacement habituel du corps calleux.
Figure 2
Coupe anatomopathologique coronale montrant une agénésie complète du corps calleux et une préservation du faisceau de Probst (flèche jaune) attaché au fornix.
Ce patient est décédé à 26 ans. Une mutation (2436delG) de l’exon 18 du gène SLC12a6 a été identifiée chez lui (cliché aimablement fourni par le Docteur Sterling Carpenter).