Résumés
Résumé
Les remaniements de la paroi bronchique, connus sous le nom de « remodelage des voies aériennes », touchent une partie des sujets asthmatiques. Ces altérations sont responsables d’un déclin progressif et irréversible de la fonction respiratoire et de la constitution d’un trouble ventilatoire obstructif irréversible et résistant aux thérapies conventionnelles, notamment aux corticoïdes. Le remodelage des voies aériennes se caractérise, entre autres, par des lésions épithéliales et une hyperplasie/hypertrophie/métaplasie des fibroblastes et des cellules musculaires lisses bronchiques. L’étude des mécanismes cellulaires et moléculaires qui gouvernent ces altérations tissulaires est l’étape indispensable à l’identification de nouvelles stratégies thérapeutiques préventives et/ou curatives.
Summary
Although asthma is classically defined as reversible airflow obstruction and often remits in younger subjects with milder disease, a proportion of asthmatics experience chronic symptoms, episodic exacerbations and persistent airway obstruction, despite the continuous use of β2-agonists, associated with high doses of inhaled/oral corticosteroids. These patients contribute to the majority of asthma costs through hospitalization, emergency visits, absence from work or school and use of medication. Although the mechanisms behind irreversible airflow obstruction in asthma are unclear, a prominent role has been attributed to persistent structural changes of the bronchial wall, defined as airway remodeling. Studies conducted on endobronchial biopsy samples have led to the histopathological characterization of these tissue alterations, which include chronic mucosal inflammation, extensive epithelial damage, collagen deposition, subepithelial fibrosis, increased mucous glands and airway smooth muscle hypertrophy and/or hyperplasia. Several factors, such as polypeptide growth factors and their receptors, matrix metalloproteases, intracellular molecules controlling cell death and survival, adhesion molecules and their ligands, as well a large variety of cytotoxic pro-inflammatory mediators are likely to contribute to the onset and maintenance of these tissue abnormalities. However, to date, the cellular and molecular events driving specifically these phenomena and allowing asthmatics with persistent airflow limitation to be distinguished from patients who normalize their bronchial obstruction upon adequate therapeutic management have not been identified yet. Accordingly, airway remodeling represents a major research challenge, particularly in view of the development of new therapeutic strategies specifically addressed at alleviating persistent bronchial obstruction in these otherwise intractable patients.
Corps de l’article
L’asthme: généralités, caractéristiques histopathologiques et traitements
L’asthme est une maladie particulièrement invalidante apparaissant généralement au cours des premières années de la vie, mais également à l’âge adulte, et qui ne bénéficie, à l’heure actuelle, d’aucun traitement curatif [1]. Différentes études épidémiologiques ont rapporté une augmentation de la prévalence de l’asthme, chez les sujets jeunes, au cours des trente dernières années, dans différents pays, dont la France [2]. Cette prévalence se situe, chez l’enfant, entre 2,1 et 32,2 %, en fonction des pays. Une définition consensuelle de l’asthme a établi qu’il s’agit une maladie chronique des voies aériennes, caractérisée par une inflammation de la paroi bronchique [3]. Cette inflammation est responsable des symptômes (dyspnée paroxystique sifflante, essoufflement, sensation d’oppression thoracique, toux) et d’une insuffisance respiratoire de degré variable qui est, au moins en partie, réversible spontanément ou à l’aide d’un traitement [3]. Le processus inflammatoire dans l’asthme se caractérise par une accumulation, dans la paroi bronchique, d’éosinophiles, de lymphocytes T de phénotype CD4+ ou CD8+, de lymphocytes T γδ et de cellules B, de macrophages, de cellules dendritiques, de mastocytes et de plaquettes [4]; la plupart de ces types cellulaires présentent des marqueurs d’activation à leur surface [4].
Le degré de sévérité de l’asthme détermine les modalités du traitement des patients. Ces traitements agissent sur la survenue des symptômes (bronchodilatateurs, agonistes de type β2-adrénergiques) et servent à limiter l’inflammation (corticoïdes) [3]. L’effet des corticoïdes, qui se traduit par une diminution des symptômes et du nombre de crises et par une amélioration de l’obstruction bronchique, semble consécutif à leurs propriétés anti-inflammatoires et immunosuppressives. En effet, une inhibition de la synthèse de différentes cytokines, chimiokines et facteurs de croissance est observée [5].
Le remodelage bronchique: caractéristiques histopathologiques dans l’asthme
Différentes études cliniques ont établi que, malgré un traitement par les corticoïdes bien conduit, entre 5 et 10 % (selon les pays) de patients asthmatiques présentent un déclin progressif et irréversible de leur fonction respiratoire [6]. À cette évolution péjorative peut se surajouter une obstruction bronchique irréversible, responsable d’une insuffisance respiratoire chronique et d’un handicap fonctionnel parfois sévère [7]. Ce sont ces cas qui rendent compte de la plus grande partie des coûts liés à l’asthme, coûts directs en rapport avec l’hospitalisation et le traitement médical, ou coûts indirects, liés pour l’essentiel à l’absentéisme scolaire ou professionnel [7]. La survenue de ces altérations fonctionnelles respiratoires serait la conséquence d’un épaississement progressif de la paroi bronchique dû à un processus de remodelage tissulaire [8].
Sur le plan histopathologique, le remodelage bronchique se caractérise par une desquamation de l’épithélium accompagnée d’une augmentation de l’espace situé entre les cellules épithéliales basales, une hypertrophie et une hyperplasie du muscle lisse, une hypertrophie des cellules glandulaires, associée à une hypersécrétion de mucus et à une fragmentation des fibres d’élastine du tissu conjonctif. Un épaississement de la membrane basale, accompagné d’une fibrose sous-épithéliale, caractérisée par un dépôt de collagène, de ténascine et de fibronectine avec une augmentation du nombre de fibroblastes et de myofibroblastes est également observé [8, 9] (Figure 1).
Figure 1
Caractéristiques histopathologiques du remodelage bronchique dans l’asthme.

Sur une coupe provenant d’une biopsie bronchique d’un sujet témoin (A), l’épithélium est intact et la muqueuse bronchique ne présente pas d’infiltrat de cellules inflammatoires, ni de fibrose sous-épithéliale. La masse musculaire est limitée. Sur des coupes issues de biopsies bronchiques de patients asthmatiques (B-E), l’épithélium est lésé (B, C et D), la membrane basale (MB) est épaissie (D), la masse musculaire est augmentée de façon notable (E) et la muqueuse bronchique est infiltrée par des éosinophiles (B), des fibroblastes et des myofibroblastes (C et D), qui sécrètent du collagène (C). Des immunomarquages ont été effectués sur des cryosections fixées en acétone en utilisant des anticorps reconnaissant spécifiquement les éosinophiles (B), les fibroblastes et les myofibroblastes (D), le collagène de type IV (C) et les cellules musculaires lisses (E), suivis d’une révélation à la phosphatase alcaline (grossissements x 300).
Ces modifications structurales sont observées dans les bronches segmentaires et semblent spécifiques de l’asthme. Ainsi, l’analyse de prélèvements bronchiques de sujets atteints de broncho-pneumopathie chronique obstructive, une autre maladie inflammatoire pulmonaire chronique s’accompagnant d’une détérioration irréversible de la fonction respiratoire [10], montre plutôt une métaplasie épithéliale, sans épaississement de la membrane basale et un processus de fibrose qui prédomine uniquement dans les voies aériennes distales et le parenchyme pulmonaire [11].
Hypothèses concernant l’origine du remodelage bronchique
Plusieurs éléments concourent au développement du remodelage bronchique dans l’asthme (Figure 2). Un des plus étudiés est la fibrose sous-épithéliale, qui est caractérisée par une augmentation du dépôt des protéines de la matrice extracellulaire, un déséquilibre entre l’expression des métalloprotéinases qui dégradent ces protéines et leurs inhibiteurs endogènes (les TIMP), une accumulation de fibroblastes et de myofibroblastes activés, et une lyse des fibres élastiques qui contribue à rigidifier les voies aériennes [9, 11]. Cette fibrose s’accompagne d’une inflammation chronique de la muqueuse bronchique qui se manifeste notamment par une accumulation d’éosinophiles et par des lésions épithéliales persistantes, secondaires à des anomalies du processus de réparation. Enfin, une augmentation de la masse musculaire et des glandes muqueuses, accompagnée d’une hypersécretion de mucus, est fréquemment observée. Ces phénomènes sont la conséquence de phénomènes d’hypertrophie, d’hyperplasie et de métaplasie des cellules musculaires lisses et épithéliales glandulaires [9, 11].
Figure 2
Conséquences possibles d’un dérèglement de l’équilibre survie-prolifération/apoptose dans la pathogénie du remodelage bronchique.
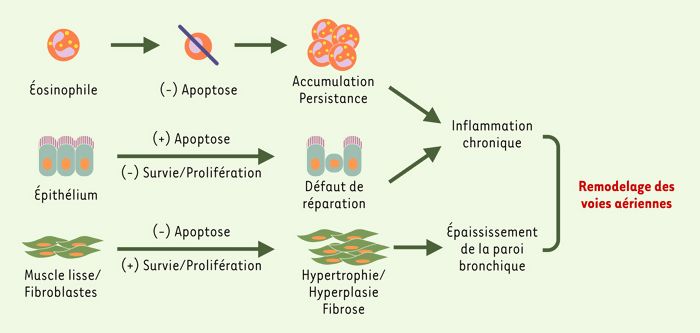
Un défaut d’élimination des éosinophiles par apoptose pourrait rendre compte de leur accumulation incontrôlée et de leur persistance dans les voies aériennes, ce qui contribuerait au maintien de l’inflammation chronique et des lésions tissulaires. En revanche, une apoptose accrue, associée à un déficit de prolifération des cellules épithéliales bronchiques, pourrait être à l’origine d’un défaut de réparation épithéliale qui perpétuerait le contact entre les allergènes transportés par voie aérienne et les structures sous-épithéliales, et entretiendrait ainsi l’inflammation et les lésions tissulaires. Enfin, une prolifération exagérée et un déficit d’apoptose des fibroblastes et des cellules musculaires lisses pourraient respectivement favoriser le développement d’une fibrose sous-épithéliale et d’une hyperplasie musculaire.
Le déséquilibre prolifération/apoptose
Le remodelage tissulaire serait le résultat de lésions répétées de la paroi bronchique, suivies d’un processus mal contrôlé de réparation tissulaire qui permet normalement l’élimination des cellules dont la fonction est altérée et leur remplacement par des cellules fonctionnelles [9]. Les lésions et la réparation anormale qui s’ensuit peuvent conduire à une métaplasie/hypertrophie/hyperplasie des différents tissus et mettent en jeu des cycles d’apoptose et/ou de nécrose, de prolifération et de différenciation des cellules inflammatoires et structurelles de la paroi bronchique, comme les cellules épithéliales, mésenchymateuses et musculaires lisses. Il a été proposé qu’un déséquilibre dans les processus qui contrôlent la survie et/ou la prolifération et l’apoptose de ces cellules (éosinophiles, cellules épithéliales, cellules musculaires lisses ou fibroblastes) puisse favoriser une inflammation chronique et le remodelage des voies aériennes dans l’asthme [12] (Figure 3). Par exemple, un défaut d’apoptose des éosinophiles pourrait rendre compte de leur accumulation incontrôlée et de leur persistance dans les voies aériennes et entretenir ainsi l’inflammation chronique et les lésions tissulaires [12-14]. Ces lésions sont engendrées notamment par les protéines cationiques granulaires, par les métalloprotéases et des facteurs fibrosants, en particulier le transforming growth factor β (TGF-β), qui exercent une action cytotoxique ou régulatrice sur la croissance et la prolifération des cellules environnantes. Inversement, une apoptose accrue, associée à un déficit de prolifération des cellules épithéliales bronchiques, pourrait être à l’origine d’un défaut de réparation épithéliale qui perpétuerait le contact entre les allergènes transportés par voie aérienne et les structures sous-épithéliales et entretiendrait ainsi l’inflammation et les lésions tissulaires [14]. Enfin, une réponse proliférative exagérée, associée à un déficit d’apoptose des fibroblastes et des cellules musculaires lisses, pourrait expliquer, du moins en partie, l’apparition d’une fibrose sous-épithéliale et l’augmentation de la masse musculaire (Figure 3).
Figure 3
Schéma récapitulatif des principales caractéristiques du remodelage des voies aériennes dans l’asthme.

D’un point de vue histopathologique, ce phénomène s’identifie par une fibrose sous-épithéliale, associée à une inflammation chronique et à une hypertrophie, hyperplasie et métaplasie de différents tissus. Ces caractéristiques sont la résultante de nombreuses modifications cellulaires à la fois morphologiques et fonctionnelles, dont des exemples sont donnés dans les encadrés. MMP: métalloprotéinases matricielles; TIMP: inhibiteurs tissulaires des métalloprotéinases.
Les anomalies morphologiques et fonctionnelles de l’épithélium et du mésenchyme
L’épithélium des voies respiratoires est le premier lieu de rencontre entre l’organisme et les particules exogènes transportées par voie aérienne et, de ce fait, un rôle clé dans les défenses et les régulations fonctionnelles des voies aériennes lui a été attribué. La cohésion de l’épithélium, et donc le maintien de sa fonction de protection et de barrière, est assurée par deux types de mécanismes: la formation des jonctions intercellulaires et les processus de réparation. Les jonctions intercellulaires comprennent les jonctions de type « serré » (présentes au pôle apical de la membrane basolatérale des cellules épithéliales ciliées et sécrétoires) et celles de type « intermédiaire » (situées sous les jonctions serrées). De plus, l’ancrage des cellules basales aux éléments du cytosquelette et de la matrice extracellulaire s’effectue par le biais des hémi-desmosomes. L’ensemble de ces jonctions est constitué de protéines différentes, comme la zonula occludens (ZO)-1, -2, -3 et l’occludine, dans le cas des jonctions serrées, les cadhérines, dans le cas des jonctions intermédiaires, et les intégrines et la laminine, qui assurent l’ancrage à la lame basale [15, 16].
Dans les voies aériennes proximales (trachée et bronches), une agression modérée est réparée grâce à la stimulation de la division des cellules sécrétoires et de leur différenciation en cellules ciliées. Lorsque l’agression est importante, la réponse fait intervenir les cellules basales qui restituent la continuité de l’épithélium en se rejoignant et en établissant entre elles des jonctions serrées. Puis, ces cellules se divisent et se différencient jusqu’au rétablissement d’un épithélium normal. Chez les sujets asthmatiques, même atteints d’asthme modéré, l’épithélium respiratoire est lésé, et cela même pendant les périodes de rémission de la maladie [4]: les cellules différenciées qui reposent sur la couche basale sont fréquemment détachées. Dans l’asthme sévère, une perte des cellules basales peut également être observée [4, 11]. Ces observations ont fait évoquer des anomalies de la réparation épithéliale à l’origine du remodelage des voies aériennes dans l’asthme [17, 18]. En effet, ces anomalies pourraient perpétuer le contact entre les allergènes et les structures sous-épithéliales et favoriser ainsi le maintien de l’inflammation tissulaire. Toutefois, les mécanismes moléculaires impliqués dans ce phénomène n’ont pas été élucidés.
Rôle des facteurs de croissance
Une des hypothèses la plus étudiée concerne une altération de l’expression et/ou de la fonction de certains facteurs de croissance, comme le keratinocyte growth factor (KGF), l’epidermal growth factor (EGF), le basicfibroblast-growth factor (bFGF) et l’hepatocyte growth factor (HGF). Il s’agit de facteurs dérivés du mésenchyme pulmonaire capables d’induire la prolifération de l’épithélium bronchique et alvéolaire [19]. En particulier, le KGF accélère la réparation de l’épithélium alvéolaire, du derme et du côlon en augmentant la prolifération et la migration cellulaire [20, 21]. D’une façon similaire, le bFGF-2 facilite la régénération épithéliale in vivo et in vitro [22]. Enfin, l’EGF est connu comme étant l’un des plus puissants agents mitogènes agissant sur les cellules épithéliales et mésenchymateuses [23]. Ce facteur règle également plusieurs autres fonctions de l’épithélium, dont la différenciation, la motilité et l’expression de molécules d’adhérence [23]. L’ajout d’EGF à des cultures cellulaires d’origine bronchique ou son application locale sur le derme favorisent la réparation épithéliale [23, 24].
Les propriétés de ces facteurs de croissance, compatibles avec leur rôle dans les processus de réparation épithéliale, ont été le plus souvent mises en évidence in vitro, sur des systèmes cellulaires ou dans des modèles animaux de fibrose pulmonaire. Ainsi, l’hypothèse d’une anomalie de l’expression et/ou de la fonction de ces facteurs dans le remodelage bronchique dans l’asthme reste à vérifier. Dans ce contexte, il est cependant important de mentionner le travail de Puddicombe et al. [24] qui ont rapporté des altérations de l’expression du récepteur de l’EGF dans les zones lésées de l’épithélium respiratoire de patients asthmatiques, et ceci en relation avec la présence d’un remodelage bronchique. Ces auteurs suggèrent que des anomalies d’expression et/ou de fonction de l’EGF et de son récepteur favoriseraient la persistance des lésions épithéliales, en empêchant le déroulement normal du processus de réparation.
De façon intéressante, tous ces facteurs de croissance et leurs récepteurs ont été impliqués dans le processus de morphogenèse du poumon au cours de l’embryogenèse [25]. Récemment, Holgate et al. [17] ont suggéré que le remodelage dans l’asthme présenterait des caractéristiques similaires aux remaniements structurels observés lors de la morphogenèse bronchique. Ce phénomène fait appel à l’activation à la fois des cellules épithéliales et de celles du mésenchyme. Cette notion a amené ces auteurs à proposer l’hypothèse selon laquelle le remodelage bronchique, et en particulier les lésions épithéliales, font suite à une réactivation de l’unité fonctionnelle épithélium-mésenchyme. Ce phénomène se traduirait par la libération, à partir de l’épithélium, de molécules pro-inflammatoires et de facteurs de croissance qui, en activant et en induisant la prolifération des cellules mésenchymateuses, contribueraient au maintien de la fibrose sous-épithéliale, une des caractéristiques du remodelage des voies aériennes (Figure 4).
Figure 4
Hypothèses concernant le rôle des relations épithélium/mésenchyme dans le remodelage des voies aériennes dans l’asthme.

La persistance des lésions épithéliales pourrait résulter d’une réactivation de l’unité fonctionnelle épithélium-mésenchyme, comme cela a été observé lors des remaniements structurels au cours de la morphogenèse bronchique. Ce phénomène se traduirait par la libération, par l’épithélium, de facteurs de croissance qui, en activant et en induisant la prolifération des cellules mésenchymateuses, contribueraient au maintien de la fibrose sous-épithéliale. Ces mêmes molécules agissent sur les cellules musculaires lisses en favorisant leur prolifération et en augmentant ainsi la masse musculaire. Il en résulte un épaississement global des voies aériennes conduisant à un rétrécissement de la lumière bronchique, source d’anomalies fonctionnelles respiratoires. Les fibroblastes et les myofibroblastes sécrètent également des facteurs de croissance responsables de la migration et de la prolifération des cellules épithéliales. Des anomalies de l’expression et/ou de la fonction de ces facteurs et de leurs récepteurs favoriseraient la persistance des lésions épithéliales, en empêchant le déroulement normal du processus de réparation. R: récepteur.
Rôle des jonctions intercellulaires
Malgré leur intérêt physiopathologique potentiel, très peu d’études ont jusqu’à présent porté sur la distribution et le rôle fonctionnel des protéines impliquées dans le maintien de la cohésion cellulaire et de l’ancrage à la matrice extracellulaire au cours des processus de lésion/réparation dans l’asthme. Ainsi, il a été montré que l’expression de CD44, un récepteur de certaines molécules de la matrice extracellulaire, est augmentée dans les zones lésées de l’épithélium bronchique des sujets asthmatiques et, in vitro, lorsque les cellules épithéliales perdent leur contact intercellulaire [17]. Bien que le rôle de CD44 dans les processus de lésion/réparation épithéliales dans l’asthme ne soit pas clair, son activation est connue pour conduire ou inhiber l’apoptose, en fonction du type cellulaire [26]. Quelques éléments d’information proviennent également d’études récentes effectuées sur la E-cadhérine. Celle molécule d’adhérence est rattachée au cytosquelette à travers l’interaction avec la β-caténine [27], protéine cytoplasmique pouvant migrer dans le noyau où elle acquiert une fonction de co-facteur de transcription [28] en réglant ainsi l’expression de nombreux gènes, dont des protéines du cycle cellulaire [29]. Comme CD44, la E-cadhérine peut régler directement l’apoptose et la prolifération cellulaire [28, 29]. Ainsi, la surexpression de cette protéine dans des lignées de cellules épithéliales et de fibroblastes humaines bloque le cycle cellulaire et induit l’apoptose [29]. Un travail récent a montré une expression amoindrie de la E-cadhérine dans l’épithélium, associée à une libération de sa forme soluble dans le liquide de lavage broncho-alvéolaire, dans un modèle d’asthme allergique chez le cobaye [30]. Ces phénomènes s’accompagnant d’une augmentation de la perméabilité épithéliale, il a été suggéré que le clivage de la E-cadhérine, témoin d’une perte de l’intégrité épithéliale, pourrait favoriser et entretenir l’inflammation et les lésions tissulaires dans l’asthme. Une diminution de l’expression de la E-cadhérine et de la β-caténine a également été décrite in vitro, sur des cellules épithéliales bronchiques humaines stimulées avec du TNF-α [31], suggérant que des médiateurs pro-inflammatoires produits dans les voies aériennes d’asthmatiques sont responsables d’une perte de la cohésion épithéliale.
Rôle des facteurs cytotoxiques
Les lésions épithéliales dans l’asthme seraient la conséquence d’une libération locale et continue de produits cytotoxiques dérivés des éosinophiles, comme les protéines cationiques (dont la major basic protein), des enzymes protéolytiques (métalloprotéase matricielle-9) et des protéases issues des mastocytes (tryptase, chymase) [18]. Les stimulus environnementaux, comme les allergènes, les bactéries ou les virus peuvent également provoquer des lésions épithéliales, notamment par le biais d’une activité protéasique [18]. Ces lésions épithéliales se traduisent non seulement par des changements structuraux, mais également par des modifications phénotypiques et fonctionnelles. Celles-ci incluent une augmentation de l’expression de molécules d’adhérence, de la libération de médiateurs lipidiques, de cytokines, de chimiokines et de facteurs de croissance capables, à leur tour, d’activer les cellules environnantes et d’entretenir et d’amplifier l’inflammation tissulaire [18].
L’hypertrophie et l’hyperplasie du muscle lisse
L’épaississement du muscle lisse bronchique, mis en évidence notamment chez des asthmatiques décédés d’asthme aigu grave, peut être secondaire à une hypertrophie (augmentation de la taille, reflétant une synthèse protéique accrue) et/ou une hyperplasie (augmentation du nombre, conséquence d’une prolifération cellulaire excessive) des cellules musculaires lisses. Ces modifications morphologiques du muscle lisse sont associées à des altérations de l’expression de divers marqueurs membranaires et de protéines contractiles et du cytosquelette. Ces observations ont permis de proposer l’existence d’au moins deux phénotypes des cellules musculaires lisses, dits « synthétique/prolifératif » et « contractile » [32]. Ainsi, une hyperplasie musculaire serait associée à un phénotype synthétique/prolifératif. Elle est, en effet, caractérisée par une prolifération cellulaire rapide et intense, par une augmentation de l’expression de CD44, de certains facteurs de croissance et de leurs récepteurs, ainsi que des protéines constituant le cytosquelette, comme la vimentine et la desmine. De plus, une diminution de l’expression de plusieurs protéines contractiles et de leurs effecteurs, dont l’α-actine, les chaînes lourdes et légères de la myosine, la h-caldesmone, la calponine-h1, la kinase de la chaîne légère de la myosine et sa forme phosphorylée, a été observée sur ces cellules [32]. L’hypertrophie musculaire, au contraire, serait plutôt associée à un phénotype contractile qui se manifeste par un faible renouvellement cellulaire, par une augmentation importante du contenu en protéines contractiles et par une chute de l’expression de celles du cytosquelette et des facteurs de croissance [32].
Des changements du phénotype (prolifératif versus contractile) des cellules musculaires lisses ont été associés à diverses maladies caractérisées par un remodelage tissulaire. C’est le cas notamment de l’athérosclérose où les cellules musculaires lisses vasculaires présentent un phénotype prolifératif et contiennent de faibles quantités de protéines contractiles [33], alors que, dans l’insuffisance cardiaque, une hypertrophie musculaire, accompagnée d’une augmentation des protéines contractiles, a été observée [34].
Très peu d’études, en revanche, ont été consacrées à l’analyse du phénotype des cellules musculaires lisses bronchiques dans l’asthme, et ces travaux ont été menés exclusivement in vitro. Ainsi, il a été montré que l’angiotensine II favorise l’hypertrophie musculaire lisse via l’activation des facteurs de transcription c-fos et egr-1 et la libération de TGF-β. Ce facteur de croissance agirait, à son tour, de façon autocrine, en provoquant l’hypertrophie des cellules musculaires lisses bronchiques [35]. Par ailleurs, dans un modèle d’asthme allergique chez le rat, Panettieri et al. [36] ont montré une prolifération accrue des cellules musculaires lisses bronchiques, témoin de l’apparition d’une hyperplasie musculaire. Cependant, aucune étude n’a été consacrée à l’analyse du phénotype musculaire sur des prélèvements bronchiques de patients asthmatiques, en particulier en relation avec la fonction respiratoire. Ainsi, l’hypothèse selon laquelle des altérations morphologiques et phénotypiques du muscle lisse pourraient participer à l’augmentation de la masse musculaire lors du remodelage des voies aériennes, et contribuer ainsi à la dégradation de la fonction respiratoire dans l’asthme, reste à vérifier.
Le remodelage bronchique: un nouvel enjeu thérapeutique
L’inefficacité des corticoïdes observée chez 5 à 10 % des patients asthmatiques serait liée à une capacité limitée de ces composés d’inhiber certaines altérations structurales caractéristiques du remodelage bronchique comme l’épaississement de la membrane basale ou la fibrose sous-épithéliale [9, 11]. Par analogie avec les cellules mononucléées sanguines, un dérèglement de l’expression et/ou de la fonction des récepteurs des corticoïdes sur les cellules de la paroi bronchique a été proposé pour rendre compte de l’inefficacité de ce traitement [37, 38].
D’autres composés ont été testés dans le cadre d’essais cliniques. Il s’agit notamment d’immunosuppresseurs ou d’agents immunomodulateurs, comme la ciclosporine A et le méthotrexate, ou les γ-globulines [39]. Malheureusement, ces produits n’ont pas notablement amélioré l’obstruction bronchique chez ces patients, suggérant une action soit mal ciblée, soit tardive alors que les altérations tissulaires et fonctionnelles respiratoires étaient déjà établies.
Des études, à des stades plus ou moins avancés de développement, font espérer l’émergence de nouvelles thérapeutiques permettant, d’une part, de limiter les effets secondaires des traitements au long cours par les corticoïdes et, d’autre part, de prendre en charge les sujets présentant une résistance à ces traitements. Les molécules potentiellement efficaces sont actuellement à l’étude: elles agissent sur différentes cibles cellulaires ou moléculaires impliquées dans le remodelage bronchique et plus particulièrement dans la survenue de la fibrose. Un anticorps humanisé dirigé contre les immunoglobulines E a été développé; il bloque l’établissement de l’inflammation dès la réaction précoce allergique et limite ainsi le recours aux corticoïdes [40]. Les antagonistes des cystéinyl-leucotriènes pourraient constituer une option thérapeutique complémentaire ou alternative aux corticoïdes. En effet, les cystéinyl-leucotriènes, médiateurs lipidiques, augmentent la prolifération des cellules épithéliales et des cellules musculaires lisses bronchiques et elles induisent l’expression et l’activation de certaines protéases de la matrice extracellulaire. Les antagonistes des récepteurs A de l’endothéline (ETA) pourraient également avoir un effet clinique: l’endothéline-1 (puissant peptide bronchoconstricteur) stimule la sécrétion de collagène par les fibroblastes pulmonaires, augmente la synthèse de fibronectine par les cellules épithéliales bronchiques humaines et amplifie la prolifération des cellules musculaires lisses bronchiques, des effets qui mettent en jeu le récepteur ETA [41, 42]. Si ces observations suggèrent une implication de l’endothéline-1 dans le remodelage bronchique, l’effet des antagonistes de l’ETA reste à tester. Enfin, il a été récemment montré que des inhibiteurs des phosphodiestérases 4 pouvaient moduler, in vitro, la dégradation de la matrice extracellulaire par les fibroblastes [43, 44], suggérant que ces composés possèderaient des propriétés anti-fibrosantes compatibles avec leur utilisation dans la prévention ou le traitement du remodelage bronchique dans l’asthme.
Aujourd’hui, l’absence d’une thérapeutique efficace peut s’expliquer par la complexité et la multiplicité des événements cellulaires et moléculaires impliqués dans ces remaniements tissulaires, mais également par la variabilité dans le temps de l’apparition de ces anomalies tissulaires au cours de l’évolution de la maladie asthmatique. Ces considérations soulignent l’importance d’identifier des marqueurs cellulaires et moléculaires prédictifs, de préférence lorsque la fonction respiratoire est encore peu ou pas dégradée. Cette approche apparaît indispensable à l’identification de nouvelles cibles moléculaires mises en jeu lors du remodelage bronchique et à l’élaboration de traitements complémentaires aux corticoïdes destinés à une meilleure prise en charge thérapeutique de ces patients. L’épithélium respiratoire pourrait être une des cibles pour le développement de nouvelles thérapeutiques visant à prévenir les risques lésionnels dus à la sécrétion de molécules cytotoxiques et à limiter ainsi l’inflammation chronique et le remodelage tissulaire dans l’asthme.
Parties annexes
Références
- 1. Gergen PJ. Understanding the economic burden of asthma. J Allergy Clin Immunol 2001; 107: S445-8.
- 2. Neukirch F, Pin I, Knani J, et al. Prevalence of asthma and asthma-like symptoms in three French cities. Respir Med 1995; 89: 685-92.
- 3. Guidelines for the Diagnosis and Management of Asthma. Expert Panareport 2. Atlanta, Ga: National Institute of Health, May 1997. National Heart, Lung and Blood Institute Publication n° 97-4051A.
- 4. Djukanovic R, Roche WR, Wilson JW, Beasley CRW, Twentyman OP, Howarth PH. Mucosal inflammation in asthma. Am Rev Respir Dis 1990; 142: 434-57.
- 5. Barnes PJ. Mechanisms of action of glucocorticoids in asthma. Am J Respir Crit Care Med 1996; 154: 21-7.
- 6. Lange P, Parner J, Vestbo J, Schnohr P, Jensen G. A 15-year follow-up study of ventilatory function in adults with asthma. N Engl J Med 1998; 339: 1194-200.
- 7. Chung KF, Godard P. ERS task force: difficult therapy-resistant asthma. Eur Respir J 1999; 13: 1198-208.
- 8. Elias JA, Zhu Z, Chupp G, Homer RJ. Airway remodeling in asthma. J Clin Invest 1999; 104: 1001-6.
- 9. Bousquet J, Jeffery P, Busse WW, Johnson M, Vignola AM. Asthma. From bronchoconstriction to airways inflammation and remodeling. Am J Respir Crit Care Med 2000; 161: 1720-45.
- 10. Barnes PJ. Chronic obstructive pulmonary disease. N Engl J Med 2000; 343: 269-80.
- 11. Jeffery PK. Remodeling in asthma and chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2001; 164: S28-38.
- 12. Vignola AM, Chiappara G, Gagliardo R, et al. Apoptosis and airway inflammation in asthma. Apoptosis 2000; 5: 473-85.
- 13. Druilhe A, Létuvé S, Pretolani M. Eosinophil apoptosis in asthma. Pathol Biol 2000; 48: 566-73.
- 14. Pretolani M. Remodeling of the respiratory tract in asthma. Biological factors and potential physiopathological implications in the apoptosis process. Rev Mal Respir 1999; 16: S27-8.
- 15. Anderson JM. Molecular structure of tight junctions and their role in epithelial transport. News Physiol Sci 2001; 16: 126-30.
- 16. Nievers MG, Schaapveld RQ, Sonnenberg A. Biology and function of hemidesmosomes. Matrix Biol 1999; 18:5-17.
- 17. Holgate ST, Davies DE, Lackie PM, Wilson SJ, Puddicombe SM, Lordan JL. Epithelial-mesenchymal interactions in the pathogenesis of asthma. Eur Respir J 2000; 105: 193-204.
- 18. Davies DE, Polosa R, Puddicombe SM, Richter A, Holgate ST. The epidermal growth factor receptor and its ligand family: their potential role in repair and remodelling in asthma. Allergy 1999; 54: 771-83.
- 19. Thompson AB, Robbins RA, Romberger DJ, et al. Immunological functions of the pulmonary epithelium. Eur Respir J 1995; 8: 127-49
- 20. Waters CM, Savla U. Keratinocyte growth factor accelerates wound closure in airway epithelium during cyclic mechanical strain. J Cell Physiol 1999; 181: 424-32.
- 21. Takeoka M, Ward WF, Pollack H, Kamp DW, Panos RJ. KGF facilitates repair of radiation-induced DNA damage in alveolar epithelial cells. Am J Physiol 1997; 272: L1174-80.
- 22. Okada-Ban M, Thiery JP, Jouanneau J. Fibroblast growth factor-2. Int J Biochem Cell Biol 2000; 32: 263-7.
- 23. Carpenter G, Wahl MI. The epidermal growth factor family. In: Sporn MB, Roberts AB, eds. Peptide growth factors and their receptors. New York: Springer-Verlag, 1991 : 69-171.
- 24. Puddicombe SM, Polosa R, Richter A, et al. Involvement of the epidermal growth factor receptor in epithelial repair in asthma. FASEB J 2000; 14: 1362-74.
- 25. Warburton D, Schwarz M, Tefft D, Flores-Delgado G, Anderson KD, Cardoso WV. The molecular basis of lung morphogenesis. Mech Dev 2000; 92: 55-81.
- 26. Johnson P, Maiti A, Brown KL, Li R. A role for the cell adhesion molecule CD44 and sulfation in leukocyte-endothelial cell adhesion during an inflammatory response? Biochem Pharmacol 2000; 59: 455-65.
- 27. Ilyas M, Tomlison IP. The interactions of APC, E-cadherin and β-catenin in tumour development and progression. J Pathol 1997; 182: 128-37.
- 28. Kikuchi A. Regulation of β-catenin signaling in the Wnt pathway. Biochem Biophys Res Commun 2000; 268: 243-8.
- 29. Stockinger A, Eger A, Wolf J, Beug H, Foisner R. E-cadherin regulates cell growth by modulating proliferation-dependent β-catenin transcriptional activity. J Cell Biol 2001; 154: 1185-96.
- 30. Goto Y, Uchida Y, Nomura A, et al. Dislocation of E-cadherin in the airways epithelium during an antigen-induced asthmatic response. Am J Respir Cell Mol Biol 2000; 23: 712-8.
- 31. Carayol N, Campbell A, Vachier I, et al. Modulation of cadherin and catenin expression by tumor necrosis factor-α and dexamethasone in human bronchial epithelial cells. Am J Respir Cell Mol Biol 2002; 26: 341-7.
- 32. Hirst SJ, Walker TR, Chilvers ER. Phenotypic diversity and molecular mechanisms of airway smooth proliferation in asthma. Eur Respir J 2000; 16: 159-77.
- 33. Aikawa M, Palanisamy NS, Kuro-o M, et al. Human smooth muscle myosin heavy chain isoforms as molecular markers for vascular development and atherosclerosis. Circ Res 1993; 73: 1000-12.
- 34. Liu X, Shao Q, Dhalla NS. Myosin light chain phosphorylation in cardiac hypertrophy and failure due to myocardial infarction. J Mol Cell Cardiol 1995; 27: 2613-21.
- 35. McKay S, de Jongste JC, Saxena PR, Sharma HS. Angiotensin II induces hypertrophy of human airway smooth muscle cells: expression of transcription factors and transforming growth factor-β1. Am J Respir Cell Mol Biol 1998; 18: 823-33.
- 36. Panettieri RA, Murray RK, Eszterhs AJ, Bilgen G, Martin JG. Repeated allergen inhalations induce DNA synthesis in airway smooth muscle and epithelial cells in vivo. Am J Physiol Lung Cell Mol Physiol 1998; 274: L417-24.
- 37. Corrigan CJ, Brown PH, Barnes NC, et al. Glucocorticoid resistance in chronic asthma glucocorticoid pharmacokinetics, glucocorticoid receptor characteristics, and inhibition of peripheral-blood T-cell proliferation by glucocorticoids in vitro. Am Rev Respir Dis 1991; 144: 1016-25.
- 38. Adcock IM, Lane SJ, Brown CR, Peters MJ, Lee TH, Barnes PJ. Differences in binding of glucocorticoid receptor to DNA in steroid-resistant asthma. J Immunol 1995; 154: 3500-5.
- 39. Spector SL. Treatment of the unusually difficult asthmatic patients. Allergy Asthma Proc 1997; 18: 153-5.
- 40. Barnes PJ. Anti-IgE antibody therapy for asthma. N Engl J Med 1999; 341: 2006-8.
- 41. Barnes PJ. Endothelins and pulmonary diseases. J Appl Physiol 1994; 77: 1051-9.
- 42. Panettieri RA, Goldie RG, Rigby PJ, Eszterhas AJ, Hay DW. Endothelin-1-induced potentiation of human airway smooth muscle proliferation: an ETA receptor-mediated phenomenon. Br J Pharmacol 1996; 118: 191-7.
- 43. Burnouf C, Pruniaux MP. Recent advances in PDE4 inhibitors as immunoregulators and anti-inflammatory drugs. Curr Pharmaceutical Des 2002; 8: 1255-96.
- 44. Kohyama T, Liu XD, Zhu YK, et al. Phosphodiesterase inhibitor Cilomast inhibits fibroblasts-mediated collagen gel degradation induced by tumor necrosis factor-α. Am J Respir Cell Mol Biol 2002; 27: 487-94.
Liste des figures
Figure 1
Caractéristiques histopathologiques du remodelage bronchique dans l’asthme.
Sur une coupe provenant d’une biopsie bronchique d’un sujet témoin (A), l’épithélium est intact et la muqueuse bronchique ne présente pas d’infiltrat de cellules inflammatoires, ni de fibrose sous-épithéliale. La masse musculaire est limitée. Sur des coupes issues de biopsies bronchiques de patients asthmatiques (B-E), l’épithélium est lésé (B, C et D), la membrane basale (MB) est épaissie (D), la masse musculaire est augmentée de façon notable (E) et la muqueuse bronchique est infiltrée par des éosinophiles (B), des fibroblastes et des myofibroblastes (C et D), qui sécrètent du collagène (C). Des immunomarquages ont été effectués sur des cryosections fixées en acétone en utilisant des anticorps reconnaissant spécifiquement les éosinophiles (B), les fibroblastes et les myofibroblastes (D), le collagène de type IV (C) et les cellules musculaires lisses (E), suivis d’une révélation à la phosphatase alcaline (grossissements x 300).
Figure 2
Conséquences possibles d’un dérèglement de l’équilibre survie-prolifération/apoptose dans la pathogénie du remodelage bronchique.
Un défaut d’élimination des éosinophiles par apoptose pourrait rendre compte de leur accumulation incontrôlée et de leur persistance dans les voies aériennes, ce qui contribuerait au maintien de l’inflammation chronique et des lésions tissulaires. En revanche, une apoptose accrue, associée à un déficit de prolifération des cellules épithéliales bronchiques, pourrait être à l’origine d’un défaut de réparation épithéliale qui perpétuerait le contact entre les allergènes transportés par voie aérienne et les structures sous-épithéliales, et entretiendrait ainsi l’inflammation et les lésions tissulaires. Enfin, une prolifération exagérée et un déficit d’apoptose des fibroblastes et des cellules musculaires lisses pourraient respectivement favoriser le développement d’une fibrose sous-épithéliale et d’une hyperplasie musculaire.
Figure 3
Schéma récapitulatif des principales caractéristiques du remodelage des voies aériennes dans l’asthme.
D’un point de vue histopathologique, ce phénomène s’identifie par une fibrose sous-épithéliale, associée à une inflammation chronique et à une hypertrophie, hyperplasie et métaplasie de différents tissus. Ces caractéristiques sont la résultante de nombreuses modifications cellulaires à la fois morphologiques et fonctionnelles, dont des exemples sont donnés dans les encadrés. MMP: métalloprotéinases matricielles; TIMP: inhibiteurs tissulaires des métalloprotéinases.
Figure 4
Hypothèses concernant le rôle des relations épithélium/mésenchyme dans le remodelage des voies aériennes dans l’asthme.
La persistance des lésions épithéliales pourrait résulter d’une réactivation de l’unité fonctionnelle épithélium-mésenchyme, comme cela a été observé lors des remaniements structurels au cours de la morphogenèse bronchique. Ce phénomène se traduirait par la libération, par l’épithélium, de facteurs de croissance qui, en activant et en induisant la prolifération des cellules mésenchymateuses, contribueraient au maintien de la fibrose sous-épithéliale. Ces mêmes molécules agissent sur les cellules musculaires lisses en favorisant leur prolifération et en augmentant ainsi la masse musculaire. Il en résulte un épaississement global des voies aériennes conduisant à un rétrécissement de la lumière bronchique, source d’anomalies fonctionnelles respiratoires. Les fibroblastes et les myofibroblastes sécrètent également des facteurs de croissance responsables de la migration et de la prolifération des cellules épithéliales. Des anomalies de l’expression et/ou de la fonction de ces facteurs et de leurs récepteurs favoriseraient la persistance des lésions épithéliales, en empêchant le déroulement normal du processus de réparation. R: récepteur.