Résumés
Résumé
Il est devenu évident que les modifications post-traductionnelles des histones sont des acteurs majeurs dans les mécanismes de régulation transcriptionnelle. Il existe quatre types majeurs de modifications des histones: l’acétylation, la méthylation, la phosphorylation et l’ubiquitinylation. Les différentes combinaisons de ces modifications formeraient un code épigénétique qui, une fois lu par des domaines protéiques spécifiques, entraînerait diverses réponses à des endroits précis du génome eucaryote.
Summary
It has become clear that post-translational modifications of histones are key players in the mechanisms of transcriptionnal regulation. There are 4 major types of histone modifications: acetylation, methylation, phosphorylation and ubiquitinylation. Different combinations of these modifications would form an epigenetic code which, once read by specific protein domains, would lead to diverse responses at precise locations within the eukaryotic genome.
Corps de l’article
La régulation de la transcription des gènes chez les eucaryotes se joue en grande partie dans la chromatine. L’unité de base de ce compactage du matériel génétique est le nucléosome, où l’ADN est enroulé autour d’un octamère protéique composé de 2 copies de chacune des histones H2A, H2B, H3 et H4. Avec le concours, entre autres, de l’histone H1, des états chromatiniens encore plus condensés, allant jusqu’au chromosome mitotique, peuvent se former. Ces différents états chromatiniens modulent l’accessibilté de l’ADN aux divers processus biologiques nucléaires. Les mécanismes qui concourent à l’ouverture et à la fermeture de la chromatine deviennent de plus en plus clairs. Deux types d’activités enzymatiques peuvent être recrutées pour modifier la structure de la chromatine: celles utilisant l’énergie de l’hydrolyse de l’ATP pour altérer les liens histone-ADN à l’intérieur du nucléosome, et celles qui modifient de façon covalente certains résidus des histones, aboutissant à une chromatine plus ouverte ou plus condensée suivant le type des modifications subies[1].
Il a été proposé que les modifications post-traductionnelles des histones formaient un «code» qui dicterait l’état chromatinien et la manière de régler un gène ou un locus [2]. Une fois établi, ce code marquerait de façon épigénétique un locus soit d’une manière transitoire ponctuelle, soit de façon stable sur de grandes régions. Les principales modifications post-traductionnelles connues des histones sont: l’acétylation, la méthylation, la phosphorylation et l’ubiquitinylation. Les différentes combinaisons de ces modifications formeraient le code qui serait par la suite lu par des protéines régulatrices et serait finalement traduit en un état chromatinien particulier: actif, permissif, restrictif ou inactif. Pour ce faire, des domaines protéiques particuliers présents dans plusieurs co-régulateurs transcriptionnels jouent un rôle dans la reconnaissance spécifique de différentes modifications d’histones. Ainsi, l’état chromatinien d’une région pourrait non seulement régler son expression génique, mais aussi être maintenu tel quel à travers plusieurs générations cellulaires [3].
Cet article décrit les quatre principaux types de modifications des histones, ainsi que les dernières avancées dans la compréhension du code des histones et de son importance dans certaines des fonctions cellulaires.
L’acétylation
C’est en 1996, trente ans après la découverte qui établissait que les histones pouvaient être acétylées, que la première histone acétyltransférase (HAT) a été identifiée sous la forme d’un co-activateur transcriptionnel. Depuis, de nombreuses enzymes ont été reconnues comme ayant une activité HAT. On les classe en deux familles: les MYST (Moz, Ybf2/Sas3, Sas2, Tip60) et les GNAT (Gcn5-related N-acetyltransferase) [4]. Un certain nombre de co-activateurs transcriptionnels possèdent également une activité HAT in vitro, celle-ci n’ayant cependant pas encore été démontrée in vivo. Il existe aussi toute une famille d’enzymes, les histones désacétylases (HDAC), capable de désacétyler les lysines des histones. Les HAT et les HDAC font généralement partie de complexes multiprotéiques dont les sous-unités accomplissent des fonctions essentielles, comme l’interaction avec des facteurs transcriptionnels liés à l’ADN ou pour coupler le complexe à des promoteurs spécifiques, ou encore pour la reconnaissance du substrat chromatinien [1, 5].
Toutes les histones sont susceptibles d’être acétylées sur des lysines spécifiques dans leur domaine amino-terminal (Figure 1). La neutralisation de la charge positive de la lysine peut mener à la diminution des contacts avec l’ADN nucléosomique, avec les nucléosomes voisins et avec certaines protéines régulatrices établissant ou stabilisant l’hétérochromatine. Ces changements donnent naissance à une chromatine plus flexible, où l’ADN est plus accessible. À l’échelle cellulaire, le niveau d’acétylation délimite topographiquement l’euchromatine et l’hétérochromatine au moyen d’un gradient d’acétylation/ désacétylation [6]. L’acétylation des histones joue aussi un rôle dans la progression du cycle cellulaire, la recombinaison et la réparation de l’ADN, ainsi que dans l’apoptose [7].
Figure 1
Résidus d’acides aminés des histones sujets à des modifications post-traductionnelles chez les mammifères.

La lysine 9 de l’histone H3 peut être soit acétylée, soit méthylée, ces modifications étant mutuellement exclusives. Chez la levure Saccharomyces cerevisiae, la sérine 129 de l’histone H2A peut être phosphorylée en réponse à certains dommages de l’ADN, mais n’est pas représentée ici. Cette modification n’est présente chez les mammifères que sur le variant d’histone H2A.X, en position 139.
La méthylation
La méthylation peut survenir sur les lysines ou sur les arginines, permettant de classer les histones méthyltransférases (HMT) selon l’acide aminé modifié. Les PRMT (protein arginine methyltransferase) peuvent méthyler les arginines; les protéines à domaine SET (Su[var]3-9, enhancer of zeste, trithorax) peuvent méthyler les lysines [8]. Les lysines et les arginines ciblées sont en majorité situées sur les extrémités amino-terminales des histones H3 et H4 (Figure 1). Dot1, une HMT d’une nouvelle classe, est responsable de la méthylation d’une lysine située dans le domaine globulaire de l’histone H3 nucléosomique [9, 10]. La méthylation de lysines spécifiques joue souvent, mais non exclusivement, un rôle dans la formation de l’hétérochromatine; de son côté, la méthylation des arginines semble avoir un effet positif sur la transcription [8].
Chez les eucaryotes supérieurs, la méthylation de la lysine 9 de H3 est retrouvée dans l’hétérochromatine, alors que la méthylation de la lysine 4 de H3 est liée à l’euchromatine [2]. Il a aussi été montré que le nombre de groupements méthyles présents sur une même lysine peut mener à différents types de réponses, augmentant ainsi la complexité du code des histones [11]. Un important débat divise les chercheurs autour de la méthylation. Il a pour enjeu de déterminer si la méthylation constitue une modification permanente ou réversible, ce qui revient à se demander s’il existe des déméthylases d’histones. Cette question reste toujours sans réponse, mais plusieurs hypothèses ont été émises: l’une d’elles repose sur l’existence d’un phénomène de protéolyse des domaines amino-terminaux des histones méthylées [12].
La phosphorylation
La majorité des travaux effectués sur cette modification ont été réalisés sur la sérine 10 de l’histone H3. Chez la levure Saccharomyces cerevisiae, la phosphorylation se produit lors de l’activation transcriptionnelle et durant la condensation des chromosomes mitotiques; elle peut être réalisée respectivement par les kinases Snf1 et Ipl1/Aurora [13]. Une même phosphorylation peut donc conduire à des réponses nucléaires très différentes, ce qui est expliqué par le code des histones dans lequel la combinaison de différentes modifications entraîne une réponse spécifique.
La phosphorylation des histones joue également un rôle dans la réparation de l’ADN ayant subi des cassures doubles brins. Chez la levure, Mec1 phosphoryle la sérine 129 de l’histone H2A en réponse à certains dommages de l’ADN; le résidu 139 de l’histone variante H2A.X est ciblé par une kinase ATM/ATR dans des conditions similaires chez les mammifères [14, 15]. Cette phosphorylation établit un large domaine chromatinien autour des sites de brins sur l’ADN, et permettrait le recrutement de complexes de reconfiguration de la chromatine afin de faciliter l’accès de la machinerie de réparation à l’ADN (données non publiées). D’autres résidus des histones peuvent être phosphorylés, notamment la sérine 28 de H3, la sérine 14 de H2B, ainsi que les sérines 1 de H4 et de H2A (Figure 1). La caractérisation du rôle de ces modifications et des enzymes qui y participent est en cours.
L’ubiquitinylation
L’ajout de plusieurs copies d’ubiquitine est très souvent lié à la dégradation par le protéasome de la protéine modifiée. Cette modification des histones est particulières car elle ne met en jeu qu’une simple mono-ubiquitinylation non dégradative d’une lysine dans le domaine carboxy-terminal des histones H2A ou H2B (Figure 1). Cette modification exercerait une fonction dans la spermatogenèse, dans la réponse au stress, ainsi que dans la formation de l’hétérochromatine et la régulation de la transcription. Rad6 est l’enzyme associée à l’ubiquitinylation de l’histone H2B chez la levure [16].
Le code des histones
Les histones peuvent subir de nombreuses modifications, chaque modification pouvant influencer les autres. En effet, l’immunoprécipitation de chromatine a permis de démontrer que la phosphorylation de la sérine 10 de H3 entraîne une acétylation plus importante de la lysine 14 par Gcn5 [13]. La double modification phospho-sérine 10/acétyl-lysine 14 sur la même histone H3 est maintenant interprétée comme reflétant l’activation transcriptionnelle. De plus, d’autres expériences ont montré que la phosphorylation de la sérine 10 inhibe la méthylation de la lysine 9 de H3, et inversement, ce qui explique leurs effets transcriptionnels opposés. Il en est de même pour l’acétylation de la lysine 14 qui inhibe la méthylation de la lysine 9 (Figure2). De la même manière, les modifications du domaine amino-terminal de l’histone H4 peuvent avoir des effets régulateurs les unes sur les autres. L’acétylation des lysines 8 et 12 par le coactivateur transcriptionnel p300/CBP est augmentée lorsque l’arginine 3 est méthylée [8]. De plus, l’acétylation des lysines 5, 8, 12 et 16 inhiberait la méthylation de l’arginine 3 (Figure2). Par ailleurs, il a été suggéré que le degré d’acétylation de la lysine 16 est inversement proportionnel au degré de méthylation de la lysine 20, une modification liée à la mitose [17]. Enfin, la phosphorylation de la sérine 1 de H4 inhibe fortement l’acétylation des lysines de cette histone in vitro, alors que cet effet serait contrecarré par la méthylation de l’arginine 3 (données non publiées).
Il apparaît que ces modifications ont une influence les unes sur sur les autres, non seulement lorsqu’elles sont présentes sur une même queue d’histone, mais également en trans. En effet, un lien existe entre l’ubiquitinylation de la lysine 123 de H2B et la méthylation des lysines 4 et 79 de H3 [16]. L’ubiquitinylation de H2B in vivo par Rad6 est essentielle pour la méthylation des deux résidus de H3, à son tour requise pour la formation de l’hétérochromatine (Figure 2).
Figure 2
Interrelations régulatrices entre les différentes modifications des histones.
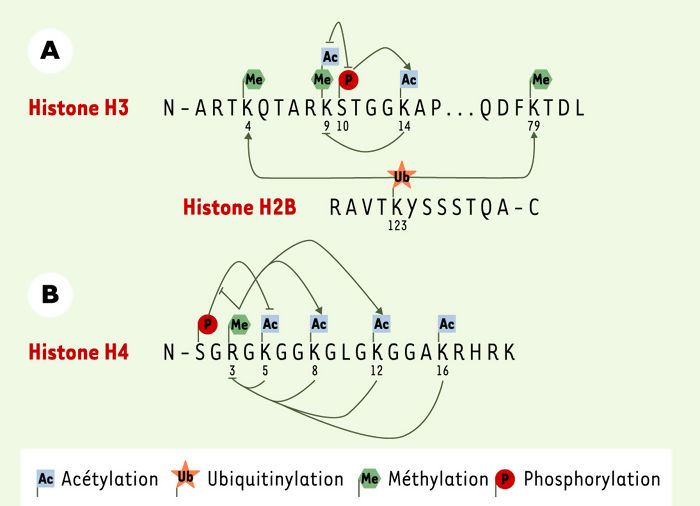
A. La phosphorylation de la sérine 10 de l’histone H3 augmente l’acétylation de la lysine 14. Ces deux modifications répriment alors la méthylation de la lysine 9. Cette méthylation inhibe la phosphorylation de la sérine 10. Chez la levure Saccharomyces cerevisiae, la mono-ubiquitinylation de la lysine 123 de l’histone H2B est nécessaire à la méthylation des lysines 4 et 79 de l’histone H3. B. La méthylation de l’arginine 3 de l’histone H4 augmente l’acétylation des lysines 8 et 12 de cette histone. En revanche, l’acétylation des résidus 5, 8, 12, et 16 inhibe la méthylation de l’arginine 3. La phosphorylation de la sérine 1 de H4 inhibe l’acétylation de la lysine 5, une inhibition bloquée par la méthylation de l’arginine 3. L’acétylation de la lysine 16 semble inhibée par la méthylation de la lysine 20, et inversement.
Le signal épigénétique porté par le locus chromatinien doit être traduit pour provoquer une réponse cellulaire. Certaines protéines responsables de cette traduction possèdent des domaines structuraux particuliers servant à la reconnaissance des différentes modifications; en outre, le plus souvent, ces protéines font partie de complexes multiprotéiques intervenant dans la régulation transcriptionnelle [2]. Les domaines les mieux caractérisés sont les bromodomaines et les chromodomaines. Il a été démontré que certains bromodomaines reconnaissent spécifiquement des lysines acétylées du domaine amino-terminal des histones et permettent ainsi la rétention de complexes protéiques sur des régions de chromatine hyper-acétylées [18]. Cela explique partiellement le recrutement séquentiel de co-activateurs transcriptionnels à des séquences régulatrices de gènes in vivo, où l’acétylation par une HAT peut être un prérequis à la mobilisation d’un complexe contenant une sous-unité à bromodomaine. Certains chromodomaines peuvent se lier à des lysines méthylées. Cette dernière interaction est très largement étudiée, car elle représente un mécanisme majeur pour la formation et la propagation de l’hétérochromatine. Le mécanisme proposé inclut la méthylation de la lysine 9 de H3 par une HMT reliée à Su(var)3-9. Une telle modification serait alors reconnue par le chromodomaine de HP1 et provoquerait sa polymérisation pour former de la chromatine très compacte sur une vaste région, l’hétérochromatine [19]. Il est intéressant de noter que cette méthylation de la lysine 9 de H3 peut diriger la méthylation de l’ADN in vivo, liant ainsi fonctionnellement ces deux mécanismes de régulation épigénétique [20].
Conclusions
Nous avons présenté différentes modifications post-traductionnelles des histones connues à ce jour. Elles suscitent un intérêt croissant, notamment celles qui concernent la formation du code épigénétique des histones, la reconnaissance de ce code par divers domaines protéiques ainsi que son impact sur les fonctions nucléaires, comme la régulation transcriptionnelle. Ce code devient de plus en plus complexe, révélant ainsi un moyen très élaboré de moduler l’expression génique de façon fine et spécifique. Ce processus fait intervenir une pléïade d’enzymes et de protéines structurales reconnaissant ces modifications grâce à des domaines spécialisés. Ces marques épigénétiques peuvent dans certains cas s’établir sur de très grandes distances, permettant à la cellule de co-régler plusieurs gènes à l’intérieur de locus chromosomiques. Ce mécanisme peut également permettre la transmission de l’information aux cours des divisions cellulaires [3]. La découverte initiale qui avait permis de mettre en évidence les liens associant les histones acétylées aux régions transcriptionnellement actives du génome est confirmée. Cependant, de nombreuses précautions doivent être prises lorsqu’on regarde le code des histones dans son ensemble. Chaque type de modification ne concerne plus exclusivement l’activation ou la répression de la transcription. Le contexte global des différentes modifications reste encore difficile à saisir car, même si une grande partie des modifications est désormais connue, il faut encore caractériser les enzymes responsables de ces modifications et étudier toutes leurs interactions et interdépendances.
Parties annexes
Remerciements
Les travaux de notre laboratoire sont subventionnés par les Instituts de recherche en santé du canada (IRSC) et la Société de recherche sur le cancer Inc. N.L. est un étudiant français boursier du ministère canadien des Affaires étrangères et du commerce international. J.C. est un chercheur des IRSC.
Références
- 1. Narlikar GJ, Fan HY, Kingston RE. Cooperation between complexes that regulate chromatin structure and transcription. Cell 2002; 108: 475-87.
- 2. Jenuwein T, Allis CD. Translating the histone code. Science 2001; 293: 1074-80.
- 3. Turner BM. Cellular memory and the histone code. Cell 2002; 111: 285-91.
- 4. Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annu Rev Biochem 2001; 70: 81-120.
- 5. Utley RT, Côté J. The MYST family of histone acetyltransferases. Curr Top Microbiol Immunol 2002; 274: 203-36.
- 6. Suka N, Luo K, Grunstein M. Sir2p and Sas2p opposingly regulate acetylation of yeast histone H4 lysine 16 and spreading of heterochromatin. Nat Genet 2002; 32: 378-83.
- 7. Carrozza MJ, Utley RT, Workman JL, Côté J. The diverse functions of histone acetyltransferase complexes. Trends Genet 2003; 19: 321-9.
- 8. Zhang Y, Reinberg D. Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev 2001; 15: 2343-60.
- 9. Khan AU, Hampsey M. Connecting the DOTs: covalent histone modifications and the formation of silent chromatin. Trends Genet 2002; 18: 387-9.
- 10. Lacoste N, Utley RT, Hunter JM, Poirier GG, Côté J. Disruptor of telomeric silencing-1 is a chromatin-specific histone H3 methyltransferase. J Biol Chem 2002; 277: 30421-4.
- 11. Santos-Rosa H, Schneider R, Bannister AJ, et al. Active genes are tri-methylated at K4 of histone H3. Nature 2002; 419: 407-11.
- 12. Bannister AJ, Schneider R, Kouzarides T. Histone methylation: dynamic or static? Cell 2002; 109: 801-6.
- 13. Berger SL. Histone modifications in transcriptional regulation. Curr Opin Genet Dev 2002; 12: 142-8.
- 14. Downs JA, Lowndes NF, Jackson SP. A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature 2000; 408: 1001-4.
- 15. Redon C, Pilch D, Rogakou E, Sedelnikova O, Newrock K, Bonner W. Histone H2A variants H2AX and H2AZ. Curr Opin Genet Dev 2002; 12: 162-9.
- 16. Briggs SD, Xiao T, Sun ZW, et al. Gene silencing: trans-histone regulatory pathway in chromatin. Nature 2002; 418: 498.
- 17. Rice JC, Nishioka K, Sarma K, Steward R, Reinberg D, Allis CD. Mitotic-specific methylation of histone H4 Lys 20 follows increased PR- Set7 expression and its localization to mitotic chromosomes. Genes Dev 2002; 16: 2225-30.
- 18. Hassan AH, Prochasson P, Neely KE, et al. Function and selectivity of bromodomains in anchoring chromatin- modifying complexes to promoter nucleosomes. Cell 2002; 111: 369-79.
- 19. Grewal SI, Elgin SC. Heterochromatin: new possibilities for the inheritance of structure. Curr Opin Genet Dev 2002; 12: 178-87.
- 20. Lachner M. Epigenetics: SUPERMAN dresses up. Curr Biol 2002; 12: R434-6.
Liste des figures
Figure 1
Résidus d’acides aminés des histones sujets à des modifications post-traductionnelles chez les mammifères.
La lysine 9 de l’histone H3 peut être soit acétylée, soit méthylée, ces modifications étant mutuellement exclusives. Chez la levure Saccharomyces cerevisiae, la sérine 129 de l’histone H2A peut être phosphorylée en réponse à certains dommages de l’ADN, mais n’est pas représentée ici. Cette modification n’est présente chez les mammifères que sur le variant d’histone H2A.X, en position 139.
Figure 2
Interrelations régulatrices entre les différentes modifications des histones.
A. La phosphorylation de la sérine 10 de l’histone H3 augmente l’acétylation de la lysine 14. Ces deux modifications répriment alors la méthylation de la lysine 9. Cette méthylation inhibe la phosphorylation de la sérine 10. Chez la levure Saccharomyces cerevisiae, la mono-ubiquitinylation de la lysine 123 de l’histone H2B est nécessaire à la méthylation des lysines 4 et 79 de l’histone H3. B. La méthylation de l’arginine 3 de l’histone H4 augmente l’acétylation des lysines 8 et 12 de cette histone. En revanche, l’acétylation des résidus 5, 8, 12, et 16 inhibe la méthylation de l’arginine 3. La phosphorylation de la sérine 1 de H4 inhibe l’acétylation de la lysine 5, une inhibition bloquée par la méthylation de l’arginine 3. L’acétylation de la lysine 16 semble inhibée par la méthylation de la lysine 20, et inversement.