Corps de l’article
Les flavivirus (famille Flaviviridae) sont des arbovirus (arthropod-borne virus) véhiculés principalement par les moustiques ou les tiques hématophages. Ce sont de petits virus enveloppés ayant pour génome un ARN simple brin, linéaire, de polarité positive et infectieux. Ces virus se répliquent dans le cytoplasme des cellules infectées et les cellules cibles varient selon que l’hôte est invertébré ou vertébré et selon le type de flavivirus. Ainsi, DEN et FJ ont un tropisme viscéral et TBE, SLE, WN et JE ciblent le système nerveux central (voir encadré). Chez l’homme, les flavivirus sont responsables de fièvres hémorragiques ou de méningo-encéphalites mortelles. La fièvre jaune, la dengue ((→) m/s 2002, n° 8-9, p. 816) et l’encéphalite japonaise sont les principales flaviviroses tropicales. Les autres flaviviroses humaines d’importance sont l’encéphalite de Saint Louis, l’encéphalite européenne transmise par les tiques et la fièvre du Nil occidental. La sévérité des infections à flavivirus dépend, d’une part, de la virulence des souches virales et de la compétence des vecteurs invertébrés et, d’autre part, de la constitution génétique des individus infectés. La déforestation, les perturbations climatiques, l’intensification des échanges internationaux, l’urbanisation anarchique, l’absence de surveillance efficace des vecteurs invertébrés et leur résistance aux insecticides sont les principaux facteurs qui favorisent l’émergence, ou la résurgence, des flaviviroses humaines dans les différentes régions du globe. La lutte anti-vectorielle reste peu efficace car elle doit associer une destruction conjointe des gîtes larvaires et des moustiques adultes.
Le virus du Nil occidental a été isolé pour la première fois en Ouganda en 1937. Il se maintient dans la nature grâce à un cycle faisant intervenir principalement les oiseaux comme réservoirs et les moustiques hématophages du genre Culex comme vecteurs [1, 2]. Les oiseaux migrateurs virémiques assurent le transport du virus dans des régions éloignées où ils vont de nouveau le transmettre aux moustiques ornithophiles du genre Culex. Beaucoup d’espèces de mammifères sont permissives au virus du Nil occidental. Les chevaux, comme les autres mammifères, sont infectés lors d’une piqûre par un moustique hématophage porteur du virus, et sont particulièrement sensibles à la maladie, mais ils ne participent pas au cycle de transmission. La fièvre du Nil occidental est une zoonose endémique en Afrique, en Asie, en Europe et en Australie. Les études phylogéniques ont révélé l’existence de deux lignées de virus : la lignée virale 1 qui a une distribution mondiale et la lignée virale 2 qui est essentiellement africaine. La lignée 1 a été responsable d’enzooties en Roumanie (1996), en Russie (1999), en Israël (1998-2000) et récemment dans l’hémisphère ouest à partir des États-Unis (1999). Les souches virales isolées lors des récentes épidémies en Israël et aux États-Unis sont identiques à plus de 99,7 %. Au Moyen-Orient et en Amérique du Nord, où le virus s’est installé durablement, on observe une mortalité importante chez les oiseaux infectés notamment les Corvidae (les corbeaux !). En Amérique du Nord, à la date d’octobre 2002, on dénombrait plus de 3300 sujets infectés par le virus du Nil occidental dont 182 décès. Actuellement, la zoonose sévit dans la plupart des régions des États-Unis (Figure 1). Ce sont principalement les moustiques du genre Culex, trouvés aussi dans les zones urbaines, qui assurent la propagation locale du virus, mais ce sont probablement des oiseaux virémiques plus ou moins résistants à l’infection virale qui vont assurer la propagation dans des régions éloignées. Normalement les mammifères, comme les chevaux et les humains, ne sont pas des amplificateurs ou des réservoirs de virus.
Figure 1
Répartition de l’infection par le virus du Nil occidental aux Etats-Unis.
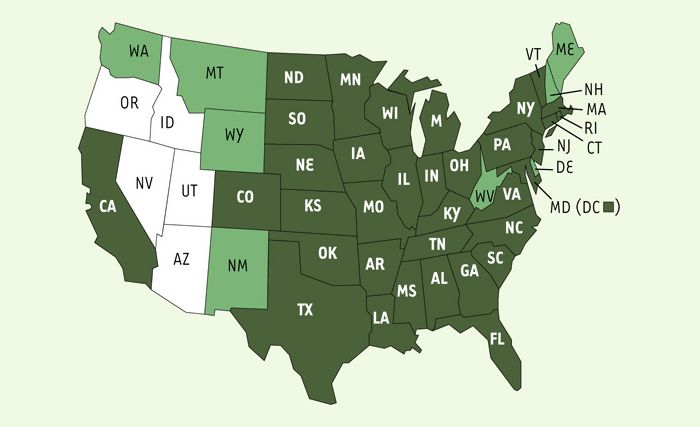
Les États dans lesquels une infection prouvée biologiquement a été détectée chez les oiseaux, les moustiques les animaux ou les humains sont colorés en vert (clair ou foncé). Les États où des cas d’infection humaine sont répertoriés sont indiqués en vert foncé.
Dans les régions tempérées et sub-tropicales, les infections humaines peuvent survenir pendant la période automnale. Chez un sujet piqué par un moustique infecté, la période d’incubation est d’environ une semaine, mais moins de 20 % des personnes infectées par le virus du Nil occidental ont des manifestations cliniques. Dans sa forme bénigne, l’infection virale se manifeste par un état fébrile associé à une faiblesse musculaire, à des céphalées et à des douleurs abdominales. Chez moins de 1 % des sujets infectés, une méningite aiguë aseptique ou une encéphalite peut survenir. D’autres manifestations - splénomégalie, hépatite, pancréatite ou myocardite - peuvent également être observées. Des paralysies flasques comme celles que l’on observe au cours d’une poliomyélite ont été récemment décrites, mais les cas mortels d’encéphalite virale (qui concernent 5 % des patients ayant des troubles neurologiques sévères) se rencontrent principalement chez les sujets fragiles et les personnes âgées [3, 4]. Chez les oiseaux infectés, on retrouve des foyers de cellules infectées par le virus dans les viscères et le cerveau. Pour l’homme, rien n’est clair. La transmission inter-humaine du virus a été récemment observée aux États-Unis chez des sujets ayant subi une transplantation d’organe ou ayant été perfusés avec des produits sanguins contaminés.
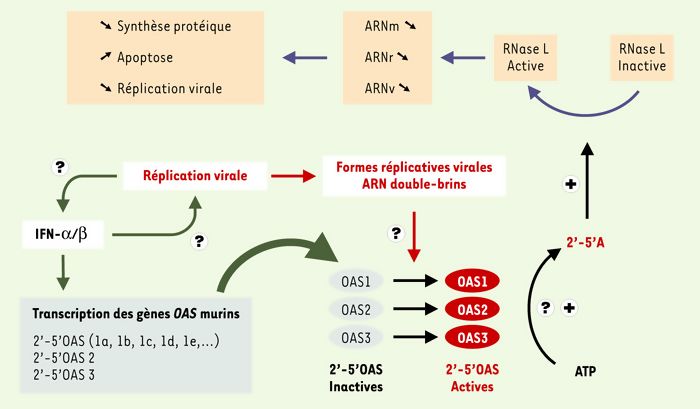
Figure 2
Système de défense inné 2’-5’ OAS/RNase L et réplication des virus à ARN.
Les souris de laboratoire peuvent être infectées expérimentalement par les flavivirus soit par inoculation intracrânienne soit, dans certains cas, par inoculation intrapéritonéale. On constate alors que la majorité des lignées consanguines de laboratoire sont sensibles à l’infection et succombent en une dizaine de jours des conséquences d’une encéphalite, tandis que d’autres lignées, dérivées de géniteurs récemment capturés à l’état sauvage et appartenant à des espèces voisines du même genre Mus résistent. Cette différence de comportement, connue depuis plusieurs dizaines d’années, relève d’un déterminisme génétique relativement simple puisqu’un seul locus désigné Flv (pour flavivirus), porté par le chromosome 5, est en cause avec au moins deux allèles : Flvr, qui confère la résistance et Flvs qui confère la sensibilité [5]. Cette sensibilité au flavivirus portée par le locus Flv est générale pour l’ensemble des flavivirus.
En réalisant des croisements entre souris sensibles et souris résistantes, nous avons identifié un gène candidat pour cette différence de sensibilité dans un segment chromosomique contenant une douzaine de gènes [6, 7]. Ce gène code pour une enzyme essentielle dans la mise en oeuvre des mécanismes innés de défense cellulaire contre une infection virale [8] : la 2’-5’-oligo adénylate synthétase (OAS) qui représente une famille de protéines induites par l’interféron α/β [9]. Lorsque l’enzyme OAS est activée par des ARN double-brins (souvent d’origine virale), elle polymérise l’ATP pour synthétiser des oligomères [pppan (2’p5A)n (1≤n≤30)] appelés 2-5A. Les molécules 2-5A activent une forme latente de l’endoribonucléase RNase L qui, une fois active, va dégrader les molécules d’ARN simple brin, cellulaires ou viraux, bloquant ainsi la réplication intracellulaire du virus (Figure 2). Par ailleurs, les gènes OAS (il en existe 10 chez la souris qui peuvent se diviser en 3 familles : OAS1, OAS2 et OAS3) sont impliqués dans la stabilité des ARNm et dans des processus physiologiques comme la division cellulaire, la différenciation et l’apoptose [8, 9]. Chez les souris sensibles, nous avons découvert que le gène qui code pour l’isoforme 2’-5’-OAS 1b était muté et présentait un codon stop dans son exon 4. Cette mutation non-sens, sans doute d’origine récente, existe chez la plupart des souris de laboratoire que nous avons étudiées mais ne se rencontre pas à l’état sauvage. Elle aboutit à la synthèse d’une molécule tronquée, sans doute non fonctionnelle et par conséquent incapable de stimuler la synthèse de la RNase L. In fine, cela aboutit à l’incapacité de se défendre pour les cellules infectées. On ne sait pas encore si une mutation de ce type existe ou non chez l’homme où le même mécanisme de défense existe aussi mais, si elle existe, ou s’il existe des allèles mutants de la 2’-5’-OAS dont le produit est moins actif que la protéine native, il est probable que ces mutations confèrent elles aussi une sensibilité particulière aux flavivirus de toute nature.
Dans l’état actuel des observations, la mutation stop que nous avons observée apparaît comme la cause probable de la sensibilité à l’infection au virus du Nil occidental mais il faut le démontrer de manière non ambiguë. Ce ne sera pas chose très facile à faire car la souris, contrairement à l’homme, possède plusieurs isoformes structurellement très voisines du gène OAS et toutes les cellules embryonnaires que nous pourrions modifier in vitro sont porteuses du fameux codon stop. Quoi qu’il en soit, une retombée importante de cette observation est qu’elle nous fournit l’occasion d’étudier dans le détail un mécanisme cellulaire de défense inné très archaïque mais déterminant dans le degré de sévérité des infections à flavivirus.

Parties annexes
Références
- 1. Brinton MA. The molecular biology of West Nile virus : a new invader of the Western hemisphere. Annu Rev Microbiol 2002 ; 56 : 371-402.
- 2. Campbell G, Marfin AA, Lanciotti RB, Gubler DJ. West Nile virus. Lancet 2002 ; 2 : 519-29.
- 3. Glass JD, Samuels O, Rich MM. Poliomyelitis due to West Nile virus. N Engl J Med 2002 ; 347 : 1280-1.
- 4. Petersen LR, Roehrig JT, Hughes JM. West Nile virus encephalitis. N Engl J Med 2002 ; 347 : 1225-6.
- 5. Perelygin AA, Scherbik SV, Zhulin IB, Stockman BM, Li Y, Brinton MA. Positional cloning of the murine flavivirus resistance gene. Proc Natl Acad Sci USA 2002 ; 99 : 9322-7.
- 6. Mashimo T, Lucas M, Simon-Chazottes D, et al. A nonsense mutation in the gene encoding 2’-5’-oligoadenylate synthetase/L1 isoform is associated with West Nile virus susceptibility in laboratory mice. Proc Natl Acad Sci USA 2002 ; 99 : 11311-6.
- 7. Samuel CE. Host genetic variability and West Nile virus susceptibility. Proc Natl Acad Sci USA 2002 ; 99 : 11555-7.
- 8. Kakuka S, Shibata S, Iwakura Y. Genomic structure of the mouse 2’-5’-oligoadenylate synthetase gene family. J Interferon Cytokine Res 2002 ; 22 : 981-93.
- 9. Samuel CE. Antiviral actions of interferons. Clin Microbiol Rev 2001 ; 14 : 778-809.
Liste des figures
Figure 1
Répartition de l’infection par le virus du Nil occidental aux Etats-Unis.
Les États dans lesquels une infection prouvée biologiquement a été détectée chez les oiseaux, les moustiques les animaux ou les humains sont colorés en vert (clair ou foncé). Les États où des cas d’infection humaine sont répertoriés sont indiqués en vert foncé.
Figure 2
Système de défense inné 2’-5’ OAS/RNase L et réplication des virus à ARN.