Corps de l’article
L’enveloppe nucléaire sépare le nucléoplasme du cytoplasme et organise la structure du noyau dans toutes les cellules eucaryotes. Elle est formée de deux membranes concentriques percées de place en place par des pores nucléaires, qui permettent à certaines molécules d’être activement transportées depuis et vers le cytoplasme. La membrane nucléaire externe est en continuité physique avec le réticulum endoplasmique dont elle partage les fonctions. La membrane nucléaire interne est, quant à elle, associée à un réseau de protéines, la lamina nucléaire, qui est interposé entre la membrane et la chromatine [1]. Cette lamina nucléaire est essentiellement composée des lamines, protéines formant des filaments intermédiaires, et elle interagit avec un certain nombre de protéines ancrées à la membrane nucléaire interne (émerine, LAP2, LBR).
La lamina nucléaire fait aujourd’hui l’objet de nombreuses recherches. Quels sont ses partenaires dans le noyau? Quels rôles jouent l’ensemble de ces protéines dans la résistance mécanique de l’enveloppe nucléaire, l’organisation du noyau et la régulation du cycle cellulaire? Le rôle de la lamina varie-t-il selon le type cellulaire? L’intérêt des chercheurs a été aiguisé par la découverte récente d’un lien entre des mutations de certaines protéines de l’enveloppe nucléaire et des maladies héréditaires. En particulier, il a été montré que la dystrophie musculaire d’Emery-Dreifuss (EDMD) est due à des mutations soit du gène codant pour l’émerine [2], soit du gène codant pour des lamines de type A (lamines A et C) [3]. Le gène codant pour des lamines de type A est également muté dans cinq autres pathologies: (1) deux pathologies de type musculaire, la dystrophie musculaire des ceintures de type IB et la cardiomyopathie dilatée [3]; (2) une pathologie du tissu adipeux, la lipodystrophie de type Dunningan [3]; (3) une pathologie affectant le développement osseux, la dysplasie mandibuloacrale [4]; et (4) une pathologie du nerf périphérique, la maladie de Charcot Marie Tooth 2 [5].
Afin de mieux comprendre l’impact des mutations qui provoquent des pathologies héréditaires sur la structure et la fonction des protéines de l’enveloppe nucléaire, notre groupe s’intéresse à la caractérisation structurale de ces protéines par les approches suivantes: (1) détermination de la structure en solution des protéines dans leurs formes natives et mutées; (2) évaluation de l’impact des mutations sur la stabilité de ces protéines; (3) cartographie des surfaces d’interaction des protéines avec leurs partenaires biologiques; (4) positionnement des mutations par rapport à ces surfaces fonctionnellement importantes; et (5) identification de la structure tridimensionnelle des complexes protéiques auxquels participent des protéines de l’enveloppe nucléaire. Ce travail est mené en étroite collaboration avec des généticiens impliqués dans l’identification des mutations provoquant les différentes pathologies (équipe de Gisèle Bonne, Institut de Myologie, Hôpital Pitié-Salpêtrière, Paris) et des biologistes cellulaires qui observent l’impact de ces mutations sur l’organisation et le fonctionnement des noyaux des cellules (équipes de Jean-Claude Courvalin, Institut Jacques Monod, Paris, France, et d’Howard J. Worman, Columbia University, New York, USA) [6]-[7]-[8].
Récemment, nous avons publié la structure tridimensionnelle en solution de la région C-terminale des lamines de type A [9]. La détermination de cette structure a été réalisée à partir de données expérimentales (valeurs des distances inter-proton inférieures à 6 Å) obtenues par résonance magnétique nucléaire du proton, de l’azote et du carbone. Des calculs de modélisation moléculaire ont permis d’identifier une famille de structures tridimensionnelles, toutes très proches les unes des autres, et compatibles avec l’ensemble des données expérimentales. Ces structures, qui reflètent la structure de la protéine en solution, présentent un repliement de type immunoglobuline entre les résidus 430 et 545. La majorité des mutations liées à la dystrophie d’Emery-Dreifuss et à d’autres pathologies de type musculaire touchent des résidus du coeur hydrophobe du domaine de type immunoglobuline (Figure 1A). Ces mutations déstabilisent probablement la structure tridimensionnelle de ce domaine, induisant une désorganisation des filaments de lamine et donc une perte globale de la fonction protéique. Cette déstabilisation a été vérifiée expérimentalement par dichroïsme circulaire sur le mutant Arg453Trp, le plus fréquemment retrouvé chez les patients atteints de laminopathies. En revanche, la majorité des mutations responsables de la lipodystrophie de type Dunnigan, une pathologie des tissus adipeux, est située dans une région du domaine de type immunoglobuline accessible aux solvants et chargée positivement (Figure 1B). Ces mutations ne déstabilisent pas la structure du domaine, mais affectent probablement l’interaction de la lamine avec l’un de ses partenaires biologiques.
Figure 1
Localisation des mutations responsables de maladies génétiques au sein du domaine de type immunoglobuline des lamines de type A.
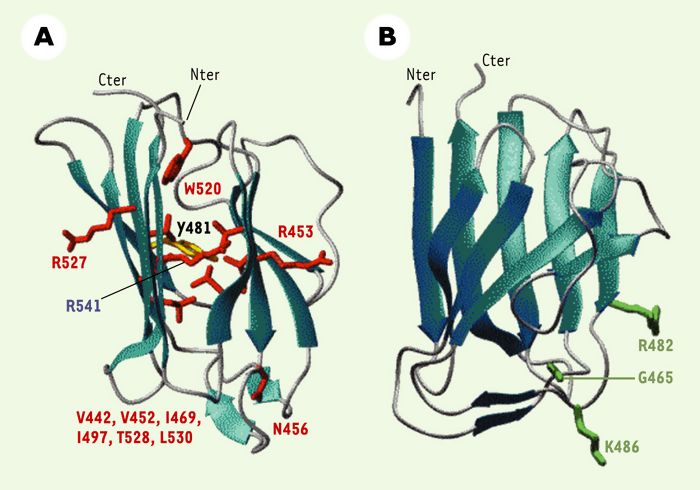
A. Maladies des muscles cardiaques et squelettiques. B. Maladies du tissu adipeux. Les mutations associées à la dystrophie musculaire d’Emery-Dreifuss sont en rouge, à la dystrophie musculaire des ceintures de type 1B en jaune (Y481), et à la lipodystrophie de type Dunnigan en vert. Le résidu arginine 541 est muté dans deux pathologies: la dystrophie musculaire d’Emery-Dreifuss (Arg541His) et la cardiomyopathie dilatée (Arg541Cys).
Il reste à présent à identifier les surfaces d’interaction de la région C-terminale de la lamine A/C avec ses partenaires biologiques et à déterminer la position relative des mutations responsables des laminopathies par rapport à ces surfaces d’interaction. Il est aujourd’hui proposé que les lamines jouent un rôle dans la réplication de l’ADN, l’organisation de la chromatine, l’arrangement spatial des pores nucléaires et des protéines ancrées dans la membrane nucléaire, et la croissance du noyau [10], [11]. Dans ce contexte, il a été montré que les lamines interagissent avec l’ADN, certaines protéines de la chromatine, comme des facteurs de transcription, et de la membrane interne, comme l’émerine. Il reste à comprendre comment différentes mutations au sein de la même molécule peuvent provoquer des pathologies spécifiques soit du muscle, soit du tissu adipeux par exemple [9]. On sait que l’expression de certains gènes est contrôlée par l’attachement de la chromatine à des sites particuliers sur la lamina nucléaire [10]. Les conséquences particulières de certaines mutations des lamines de type A pourrait s’expliquer par la perte de ces sites d’attachement sur la lamina nucléaire [11]. La recherche de partenaires de la lamine exprimés seulement dans certains tissus pourrait également permettre de mieux comprendre les fonctions perturbées dans les différentes laminopathies.
Parties annexes
Références
- 1. Worman HJ, Courvalin JC. The inner nuclear membrane. J Membr Biol 2000; 177: 1-11.
- 2. Bione S, Maestrini E, Rivella S, et al. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat Genet 1994; 8: 323-7.
- 3. Bonne G, Muchir A, Helbling-Leclerc A, Massart C, Schwartz K. Clinical and genetical heterogeneity of laminopathies. Acta Myologica 2001; 20: 138-44.
- 4. Novelli G, Muchir A, Sangiuolo F, et al. Mandibuloacral Dysplasia Is Caused by a Mutation in LMNA-Encoding Lamin A/C. Am J Hum Genet 2002 : 71: 426-31.
- 5. De Sandre-Giovannoli A, Chaouch M, Kozlov S, et al. Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2) and mouse. Am J Hum Genet 2002; 70: 726-36.
- 6. Bonne G, Di Barletta MR, Varnous S, et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat Genet 1999; 21: 285-8.
- 7. Vigouroux C, Auclair M, Dubosclard E, et al. Nuclear envelope disorganization in fibroblasts from lipodystrophic patients with heterozygous R482Q/W mutations in the lamin A/C gene. J Cell Sci 2001; 114: 4459-68.
- 8. Östlund C, Bonne G, Schwartz K, Worman HJ. Properties of lamin A mutants found in Emery-Dreifuss muscular dystrophy, cardiomyopathy and Dunnigan-type partial lipodystrophy. J Cell Sci 2001; 114: 14435-45.
- 9. Krimm I, Ostlund C, Gilquin B, et al. The Ig-like structure of the C-terminal domain of lamin a/c, mutated in muscular dystrophies cardiomyopathy, and partial lipodystrophy. Structure (Camb) 2002; 10: 811-23.
- 10. Stuurman N, Heins S, Aebi U. Nuclear lamins: their structure, assembly, and interactions. J Struct Biol 1998; 122: 42-66.
- 11. Cohen M, Lee KK, Wilson KL, Gruenbaum Y. Transcriptional repression, apoptosis, human disease and the functional evolution of the nuclear lamina. TrendsBiochem Sci 2001; 26: 41-7.
Liste des figures
Figure 1
Localisation des mutations responsables de maladies génétiques au sein du domaine de type immunoglobuline des lamines de type A.
A. Maladies des muscles cardiaques et squelettiques. B. Maladies du tissu adipeux. Les mutations associées à la dystrophie musculaire d’Emery-Dreifuss sont en rouge, à la dystrophie musculaire des ceintures de type 1B en jaune (Y481), et à la lipodystrophie de type Dunnigan en vert. Le résidu arginine 541 est muté dans deux pathologies: la dystrophie musculaire d’Emery-Dreifuss (Arg541His) et la cardiomyopathie dilatée (Arg541Cys).