Abstracts
Résumé
Le bon fonctionnement de la cellule dépend du contrôle fin des mécanismes transcriptionnels et traductionnels. De nombreuses études ont établi que les dérèglements transcriptionnels peuvent être à l’origine de maladies telles que le cancer, l’obésité et le diabète. Depuis une décennie, le rôle de la traduction des ARNm dans certaines de ces pathologies et son contrôle par la voie PI3K/Akt/mTOR a été démontré. La rapamycine, inhibiteur spécifique de mTOR, présente une forte activité anti-proliférative dans plusieurs types de cancer. De récentes études démontrent qu’elle pourrait potentiellement être efficace dans le traitement de l’obésité et du diabète. La rapamycine et ses analogues semblent donc destinés à un avenir prometteur.
Summary
Gene regulation by transcriptional and post-translational mechanisms is implicated in the regulation of cellular homeostasis. Transcriptional deregulation has been largely documented in the etiology of diseases such as cancer, obesity and diabetes. During the past decade, the control of translation initiation by the PI3K/Akt/mTOR pathway in the development of these pathologies has been documented. Rapamycin, a specific inhibitor of mTOR, demonstrates considerable anti-proliferative activity against numerous cancer types. Recent studies also demonstrated that rapamycin may be beneficial in the treatment of obesity and diabetes. Rapamycin and its analogs seem destined for a promising future and will help in the development of novel therapeutic strategies.
Article body
La traduction, sensible à la disponibilité en nutriments et en facteurs de croissance, représente une étape clé de l’expression génique impliquée dans la plupart des phases du métabolisme liées à la croissance cellulaire [1]. La synthèse protéique est un processus énergétique très coûteux strictement contrôlé, dès le stade d’initiation de la traduction. Ce contrôle est amorcé grâce à des facteurs d’initiation appelés eukaryotic initiation factors (eIF) [2], eIF4E étant le plus largement étudié. eIF4F permet le positionnement du complexe de pré-initiation 43S à l’extrémité 5’ de l’ARN messager (ARNm) (Figure 1), complexe qui analysera l’ARNm pour détecter l’AUG initiateur. Chez les eucaryotes, la traduction de la plupart des ARNm dépend d’une structure en 5’ appelée « coiffe » (ou cap) (m7GpppN où N représente n’importe quel nucléotide) et de la présence du complexe eIF4F composé de trois sous unités : eIF4E, eIF4A et eIF4G [2]. eIF4E est le facteur clé dans la régulation de l’initiation de la traduction : il interagit avec la coiffe, mais aussi avec eIF4G (un adaptateur entre les divers facteurs d’initiation) qui se lie à eIF4A (une ATPase-hélicase) et eIF3 qui associe la sous-unité ribosomique 43S pour former le complexe eIF4F. Les activités d’eIF4A et d’eIF4B dénaturent les structures secondaires de l’ARNm et facilitent la liaison du ribosome pour finalement déclencher la synthèse protéique (Figure 1) [2]. L’importance physiopathologique de la traduction ne fait aucun doute puisqu’une suractivation traductionnelle peut conduire au cancer, à l’obésité ou au diabète [3, 4]. Cet article se propose d’approfondir le lien entre la synthèse protéique et ses pathologies.
Figure 1
Contrôle de l’initiation de la traduction par 4E-BP.
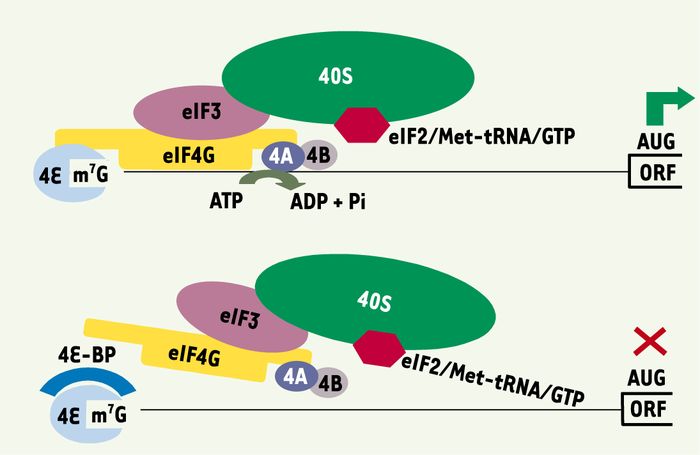
La fixation du complexe eIF4F (eIF4E, eIF4G et eIF4A) à la coiffe de l’ARNm à traduire et à la sous- unité ribosomique 40S engendre un balayage du messager qui permet la localisation du codon d’initiation AUG. L’interaction de 4E-BP avec eIF4E empêche l’interaction entre eIF4E et eIF4G et, par conséquent, la formation du complexe eIF4F. Pour leur part, eIF4A et eIF4B agissent, en tant que facteurs « accessoires », en dénaturant les structures secondaires de l’ARNm. Sous sa forme liée au GTP, eIF2 interagit avec les nucléotides guanines et l’ARN de transfert méthionine (met-tARNi). Le complexe ternaire ainsi formé (eIF2·GTP·met-tARNi) se lie à la sous unité ribosomique 40S. C’est alors l’anticodon de ce met-tARNi qui reconnaît le codon d’initiation présent dans l’ARNm. ORF : origin of replication.
Synthèse protéique et cancer
Les facteurs d’initiation de la traduction
Parmi les facteurs d’initiation de la traduction, eIF4E fut le premier impliqué dans la transformation de cellules en culture. Plusieurs ARNm, activement traduits durant la croissance cellulaire et agissant dans la transformation, contiennent des structures secondaires dans leur extrémité 5’non-traduite (vascular endothelial growth factor A, cyclin D1, fibroblast growth factor 2, ornithine decarboxylase) [3]. La surexpression d’eIF4E aboutit à un dérèglement de la synthèse protéique et du fonctionnement cellulaire (pour revue, voir [3]) et, en particulier, à une augmentation de la traduction de ces ARNm. D’autres études suggèrent également qu’eIF4E serait susceptible de coopérer avec d’autres protéines transformantes telles que c-Myc et E1A [3, 5, 6] et que son expression serait particulièrement élevée dans plusieurs types de cancer (pour revue, voir [3]). De façon similaire, le complexe eIF4F est nécessaire au maintien de la croissance des cellules cancéreuses mammaires in vivo [7]. Enfin, d’autres facteurs déclencheurs tels qu’eIF4G, eIF4A et eIF3 jouent un rôle important dans l’oncogenèse [3]. Ce constat renforce l’idée qu’une augmentation de la synthèse protéique pourrait induire l’oncogenèse.
De la signalisation à la traduction : PI3K/Akt/mTOR
Les facteurs de croissance, les agents mitogènes et les hormones activent tous la voie PI3K/Akt/mTOR et la traduction cap-dépendante [8] (Figure 2). La PI3K (phosphatidyl-inositol-3-kinase) phosphoryle le phosphatidylinositol-4,5-bisphosphate (PtdIns[4,5]P2) et produit le phosphatidylinositol-3,4,5-trisphosphate (PtdIns[3,4,5]P3). Cette production de P3 entraîne l’activation de PDK1 (3’-phosphoinositide dependent kinase 1), qui à son tour active Akt, une serine/thréonine kinase de la famille cAMP-dependent protein kinase A/ protein kinase G/ protein kinase C (AGC). Cette activation est contrôlée par PTEN, une lipide-phosphatase qui déphosphoryle les P3 et les convertit en leur forme inactive P2. PTEN est également un gène suppresseur de tumeurs souvent muté dans nombre de cancers et de syndromes associés au cancer [9]. Le complexe TSC1/TSC2 possède une activité GTPasique inhibant Rheb, une GTPase de la famille de Ras. En phosphorylant TSC2, Akt inactive le complexe TSC1/TSC2 ce qui permet à Rheb d’activer mTOR (mammalian target of rapamycin), une serine/thréonine kinase de la famille des phosphatidylinositol kinase related protein kinase (PIKK) [10]), par un mécanisme encore indéterminé [9]. TSC1 et TSC2 sont également des gènes suppresseurs de tumeurs souvent mutés dans la sclérose tubéreuse, une pathologie caractérisée par l’émergence d’hamartomes (tumeurs bénignes), principalement dans le tube digestif [9]. Lorsque le ratio AMP/ATP est élevé, l’AMPK (kinase activée par l’AMP), senseur énergétique de la cellule, phosphoryle TSC2, ce qui augmente son activité et inhibe mTOR [9]. Enfin, les acides aminés et le glucose peuvent aussi activer directement mTOR [11].
Figure 2
Intégration et régulation des signaux métaboliques par la voie PI3K/Akt/mTOR.
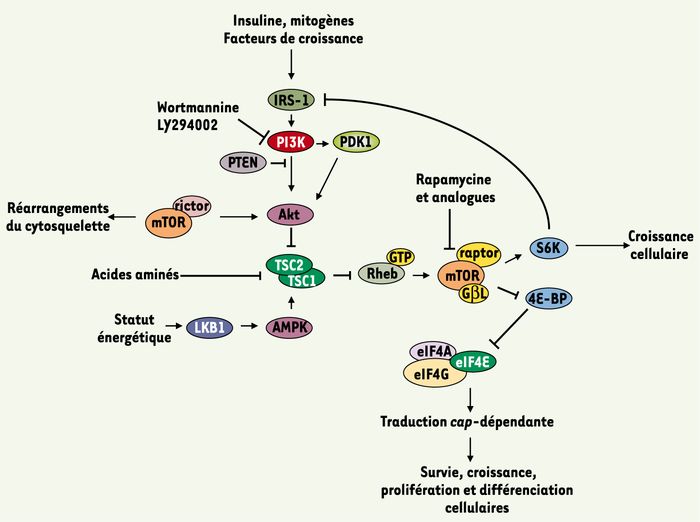
La régulation de l’activité mTOR par les facteurs de croissance fait intervenir la voie PI3K/Akt. La PI3K phosphoryle le PtdIns(4,5)P2 et produit le PtdIns(3,4,5)P3. La production de ce PtdIns(3,4,5)P3 entraîne l’activation de PDK1 qui, à son tour, active Akt. Cette activation est dépendante de PTEN. La worthmannine et le LY294002 inhibent l’activité de la PI3K. Une fois activée, cette voie de signalisation provoque la phosphorylation et l’inhibition du complexe TSC1/TSC2 par Akt, ce qui entraîne l’activation de Rheb (lié à GTP) et de mTOR. Le complexe TSC1/TSC2 joue un rôle central dans la régulation de l’activité mTOR : la présence d’acides aminés inhibe l’activité de ce complexe ; à l’inverse, une carence énergétique provoque son activation via LKB1 et AMPK. Une fois activé, le complexe mTOR/raptor/GβL, sensible à la rapamycine, relaie ces signaux vers 4E-BP et S6K. La phosphorylation de 4E-BP permet la libération d’eIF4E et la formation du complexe eIF4F nécessaire à l’initiation de la traduction. De son côté, la phosphorylation de S6K entraîne un rétrocontrôle négatif de la voie PI3K/Akt empêchant ainsi toute suractivation de la traduction cap-dépendante. L’autre complexe mTOR/rictor, insensible à la rapamycine, joue un rôle dans le réarrangement du cytosquelette.
Une fois activée, mTOR phosphoryle deux cibles majeures : les S6 kinases (S6K1 et 2) et les 4E-BP (eIF4E-binding proteins 1-3) [8] (Figure 2). L’hyperphosphorylation des 4E-BP (famille de trois protéines, 4E-BP1-3) diminue leur interaction avec eIF4E et stimule la traduction cap-dépendante (Figure 2). Les 4E-BP sont aussi d’importants régulateurs de la croissance cellulaire car la surexpression d’un mutant constitutivement actif de 4E-BP1, où les quatre sites de phosphorylation (Thr36, Thr45, Ser64 et Thr 69) sont mutés en alanines, diminue la progression en phase G1 et bloque la transformation induite par c-Myc [12]. Jusqu’à récemment, on pensait que la S6 kinase était absolument nécessaire à la traduction d’ARNm possédant une région riche en pyrimidines (TOP) dans leur partie non traduite (protéines ribosomiques eEF1α) [13]. Cette hypothèse a été réfutée en observant que chez les souris déficientes pour S6K1 et S6K2, la traduction des ARNm TOP est maintenue [14]. Cependant, la délétion de S6K1 et S6K2 mène à une réduction de la taille des souris, suggérant ainsi un nouveau rôle pour S6K dans la régulation de la croissance cellulaire [14].
mTOR permet l’intégration de nombreux signaux intra et extracellulaires et agit directement sur les cibles traductionnelles [8]. La rapamycine inhibe spécifiquement mTOR en se liant à son récepteur intracellulaire, la protéine FK506 binding protein-12 (FKBP12). Le complexe FKBP12/rapamycine ainsi formé inhibe l’activité kinase de mTOR [8]. Le développement d’analogues structuraux de la rapamycine, CCI-779 (Wyeth-Ayerst), RAD001 (Novartis) et AP23573 (Ariad Pharmaceuticals) soulève un intérêt considérable. Ces analogues présentent, en effet, une puissante activité anti-proliférative dans plusieurs types de cancer : tumeurs pulmonaires, cérébrales, mammaires, coliques, leucémies et lymphomes. Plusieurs tests cliniques faisant appel à ces analogues sont actuellement en cours de validation [15, 16].
mTOR fait partie d’un large complexe protéique comprenant trois partenaires identifiés : raptor [17, 18], GβL [19] et rictor [20, 21] (Figure 2). Raptor (regulatory associated protein of mTOR, Kog1, chez la levure), qui interagit directement avec 4E-BP et S6K grâce à leur motif TOS (séquence d’acides aminés : FEMDI et FDIDL, respectivement), est nécessaire à la phosphorylation de ces deux protéines par mTOR [22]. L’interaction raptor/mTOR est sensible à la disponibilité en nutriments et dépend d’un autre partenaire, GβL (Lst8p chez la levure) [19]. Indépendamment de raptor, GβL interagit directement avec le domaine kinase de mTOR pour augmenter son activité. Enfin, rictor (rapamycin-insensitive companion of mTOR ou mAVO3) intervient dans les réarrangements du cytosquelette [20, 21]. Rictor et raptor délimitent deux complexes mTOR : alors que l’activité du complexe mTOR/GβL/raptor (mTORC1) est inhibée par la rapamycine, le complexe mTOR/rictor (mTORC2) y est insensible [20, 21] (Figure 2). De nouvelles données font intervenir le complexe mTOR/rictor dans la phosphorylation et l’activation d’Akt [23]. Ainsi, rictor pourrait être une cible thérapeutique dans le traitement des cancers associés à la voie PI3K/Akt/mTOR [23].
Synthèse protéique, obésité et diabète
mTOR, adipogenèse et obésité
La différenciation adipocytaire est un processus complexe, chronologiquement orchestré par les facteurs de transcription C/EBP et PPARγ [24]. Dans un premier temps survient un arrêt de la prolifération des préadipocytes, puis C/EBPβ et δ sont exprimés de façon transitoire. Ces événements précoces sont suivis de l’expression de C/EBPα et PPARγ qui activent l’expression de la plupart des gènes caractérisant le phénotype adipocytaire [24] (Figure 3).
Figure 3
Régulation de l’adipogenèse et de la sensibilité à l’insuline par mTOR.
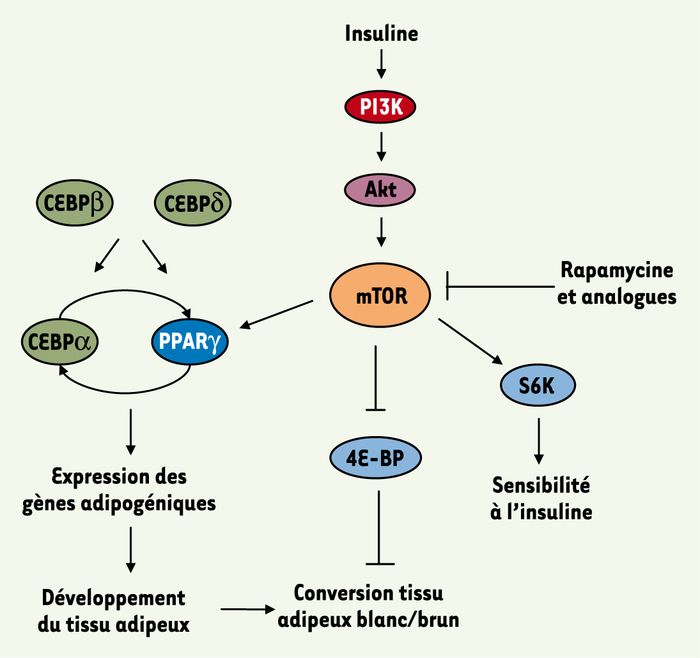
La régulation transcriptionnelle de l’adipogenèse implique l’activation de plusieurs familles de facteurs de transcription. Ceux-ci sont chronologiquement exprimés : C/EBPβ et δ, suivis de PPARγ, qui à son tour active C/EBPα. C/EBPα, via un rétrocontôle positif sur PPARγ, maintient la différenciation adipocytaire. L’adipogenèse fait également intervenir la voie mTOR. Une fois activée, elle contrôle l’expression et l’activité de PPARγ, régulant de fait l’expression des gènes adipogéniques. Via 4E-BP, mTOR module la différenciation fonctionnelle des tissus adipeux blanc et brun. Enfin, via la S6K, mTOR est également capable de réguler la sensibilité à l’insuline.
Le rôle de mTOR dans l’adipogenèse a été mis en évidence par l’utilisation de la rapamycine. Cette dernière réduit la différenciation de préadipocytes humains [25]. L’effet de la rapamycine est indépendant de C/EBPβ et C/EBPδ, mais il est associé à une réduction de l’expression de C/EBPα et PPARγ. De plus, la rapamycine réduit l’activité transcriptionnelle de PPARγ et perturbe la boucle de rétrocontrôle entre PPARγ et C/EBPα [26] (Figure 3), ce qui suggère une modulation transcriptionnelle plutôt que traductionnelle de l’adipogenèse par la rapamycine. Les rôles précis de 4E-BP1 et S6K dans le processus d’adipogenèse sont encore mal connus. La rapamycine inhibe totalement la phosphorylation de S6K, mais partiellement la différenciation adipocytaire de cellules 3T3-L1 [27] et l’expression d’un mutant constitutivement actif de S6K n’est pas capable de rétablir ce processus [27]. À l’inverse, la rapamycine n’inhibe que partiellement la phosphorylation de 4E-BP1 [27], suggérant que 4E-BP1 pourrait jouer un rôle plus important que S6K dans la différenciation adipocytaire. Il s’avère que les souris 4E-BP1-/- présentent une masse adipeuse réduite du fait d’une dépense énergétique accrue [28], associée à une conversion du tissu adipeux blanc en tissu adipeux brun, détectée par l’expression d’UCP-1 (uncoupling protein-1). De plus, PGC-1 (PPARγ∈co-activator-1), facteur de transcription impliqué dans la biogenèse mitochondriale et la thermogenèse, est également surexprimé dans le tissu adipeux blanc des souris 4E-BP1-/-. L’ensemble de ces données suggère que le contrôle de l’adipogenèse par mTOR impliquerait eIF4E/4E-BP1 plutôt que S6K. Ces données accordent également un rôle complexe à la voie mTOR par laquelle l’activation de la voie eIF4E/4E-BP1 ainsi que l’activité kinase de mTOR sont toutes deux nécessaires à l’initiation et au développement du processus adipogénique.
mTOR et insulino-résistance
Les conditions physiologiques favorisant une suractivation de la voie mTOR telles que l’hyperinsulinémie, l’excès chronique de nutriments et l’obésité, pourraient accroître l’insulino-résistance, cause de diverses maladies métaboliques, notamment le diabète de type 2 [29]. Une étude pionnière a montré qu’une exposition prolongée à l’insuline réduisait non seulement la mobilité éléctrophorétique, mais également le niveau protéique d’IRS-1 (insulin receptor substrate-1), protéine clé dans le relais des effets pléiotropiques de l’insuline [30]. Cette réduction démontrait des effets sur la transduction du signal puisque l’activation d’Akt, initialement stimulée, était réduite après une exposition prolongée à l’insuline. De plus, l’utilisation d’inhibiteurs de la voie PI3K/mTOR bloquait cet effet de l’insuline, tandis que l’utilisation d’une forme constitutivement active de la PI3K reproduisait les effets d’une exposition prolongée à l’insuline [30]. L’ensemble de ces données suggérait qu’un mécanisme mTOR-dépendant était la cause de l’inhibition de la voie PI3K/Akt via une boucle de rétrocontrôle négatif dont IRS-1 était la cible (Figure 2).
Récemment, le lien entre mTOR et le développement de l’insulino-résistance a été clarifié [29, 31, 32]. Les activités mTOR et S6K sont accrues dans le foie et le muscle squelettique de rats en régime gras [32]. Cette augmentation de l’activité mTOR est associée à une augmentation de la phosphorylation inhibitrice d’IRS-1 (Ser307, Ser636/639) et d’une inhibition d’Akt. À l’inverse, l’inhibition de mTOR permet de restaurer les activités PI3K et Akt [32]. Les délétions de TSC1 ou TSC2 sont connues pour engendrer une suractivation de la voie mTOR et de S6K [29, 31]. De façon similaire au régime gras, la suractivation de mTOR dans ces modèles réduisait l’activation d’Akt en réponse à l’insuline, à PDGF et au sérum [29, 31]. Cette inhibition de la voie Akt pouvait s’expliquer par une inhibition de l’expression d’IRS-1 [29]. Il est ici remarquable que la délétion de S6K1 corrige ce phénotype en restaurant l’activation d’Akt et l’expression d’IRS-1, suggérant que S6K1 maîtrise le rétrocontrôle négatif de la voie mTOR sur la voie PI3K/Akt [29] (Figure 2).
Il restait à déterminer si une suractivation de la voie mTOR/S6K pouvait contribuer au développement de l’insulino-résistance in vivo. L’étude de la souris S6K1-/- conforte l’idée selon laquelle S6K1 intervient dans l’inhibition d’IRS-1 [33]. Les souris témoins sous régime gras présentaient une phosphorylation accrue de S6K1 et une réduction de la phosphorylation d’Akt dans le muscle, le foie et le tissu adipeux [33]. De plus, comparées aux souris S6K1-/-, les souris témoins obèses présentaient une augmentation de la phosphorylation d’IRS-1 sur les résidus Ser307 et Ser636/639 dans le tissu adipeux, sites associés à l’insulino-résistance [34]. De la même façon, la phosphorylation de ces sites et celle de S6K1 étaient accrues chez des souris ob/ob et K/KAy, deux modèles d’obésité génétique [33].
Conclusions
La traduction est une étape essentielle dans la régulation de l’expression de nombreuses protéines nécessaires à la croissance, la prolifération et la survie cellulaire. Les données discutées dans cette suite d’analyses tendent à démontrer qu’en plus de son rôle dans le développement tumoral, une suractivation de la voie mTOR semble également jouer un rôle prépondérant dans le développement du syndrome métabolique en régulant l’adipogenèse, ainsi que la sensibilité à l’insuline, via un rétrocontrôle négatif de S6K sur IRS-1. Dans un tel contexte, une meilleure compréhension du rôle respectif des protéines 4E-BP et S6K revêt une importance clinique majeure dans le traitement du cancer, de l’obésité, du diabète, ainsi que dans le développement de nouvelles stratégies thérapeutiques. La rapamycine est, entre autres, utilisée pour réduire les risques de rejets de greffes. Les tests cliniques sur la rapamycine pour le traitement des cancers sont également porteurs de grands espoirs. Enfin, le fait que la rapamycine in vitro soit capable d’inhiber la différenciation adipocytaire et d’accroître la sensibilité à l’insuline fait de cet inhibiteur un agent thérapeutique potentiel dans le traitement de ces maladies.
Appendices
Remerciements
Les auteurs tiennent à remercier le Dr Sonenberg pour son soutien et le Dr Pelletier pour la lecture critique du manuscrit. Olivier Le Bacquer bénéficie d’une bourse postdoctorale McGill Biological Chemistry Fellowship, Yvan Martineau d’une bourse postdoctorale de l’Association pour la Recherche sur le Cancer et Yaël Mamane d’une bourse postdoctorale de l’Institut National du Cancer du Canada.
Références
- 1. Holland EC, Sonenberg N, Pandolfi PP, Thomas G. Signaling control of mRNA translation in cancer pathogenesis. Oncogene 2004 ; 23 : 3138-44.
- 2. Gingras AC, Raught B, Sonenberg N. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu Rev Biochem 1999 ; 68 : 913-63.
- 3. Mamane Y, Petroulakis E, Rong L, et al. eIF4E-from translation to transformation. Oncogene 2004 ; 23 : 3172-9.
- 4. Shi Y, Taylor SI, Tan SL, Sonenberg N. When translation meets metabolism: multiple links to diabetes. Endocrinol Rev 2003 ; 24 : 91-101.
- 5. Ruggero D, Montanaro L, Ma L, et al. The translation factor eIF-4E promotes tumor formation and cooperates with c-Myc in lymphomagenesis. Nat Med 2004 ; 10 : 484-6.
- 6. Wendel HG, De Stanchina E, Fridman JS, et al. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy.Nature 2004 ; 428 : 332-7.
- 7. Avdulov S, Li S, Michalek V, et al. Activation of translation complex eIF4F is essential for the genesis and maintenance of the malignant phenotype in human mammary epithelial cells.Cancer Cell 2004 ; 5 : 553-63.
- 8. Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev 2004 ; 18 : 1926-45.
- 9. Inoki K, Corradetti MN, Guan KL. Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet 2005 ; 37 : 19-24.
- 10. Richardson CJ, Schalm SS, Blenis J. PI3-kinase and TOR: PIKTORing cell growth. Semin Cell Dev Biol 2004 ; 15 : 147-59.
- 11. Proud CG. mTOR-mediated regulation of translation factors by amino acids. Biochem Biophys Res Commun 2004 ; 313 : 429-36.
- 12. Lynch M, Fitzgerald C, Johnston KA, et al. Activated eIF4E-binding protein slows G1 progression and blocks transformation by c-myc without inhibiting cell growth. J Biol Chem 2004 ; 279 : 3327-39.
- 13. Meyuhas O. Synthesis of the translational apparatus is regulated at the translational level. Eur J Biochem 2000 ; 267 : 6321-30.
- 14. Pende M, Um SH, Mieulet V. S6K1(-/-)/S6K2(-/-) mice exhibit perinatal ethality and rapamycin-sensitive 5’-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway.Mol Cell Biol 2004 ; 24 : 3112-24.
- 15. Rao RD, Buckner JC, Sarkaria JN. Mammalian target of rapamycin (mTOR) inhibitors as anti-cancer agents. Curr Cancer Drug Targets 2004 ; 4 : 621-35.
- 16. Dutcher JP. Mammalian target of rapamycin (mTOR) inhibitors. Curr OncolRep 2004 ; 6 : 111-5.
- 17. Kim DH, Sarbassov DD, Ali SM, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002 ; 110 : 163-75.
- 18. Hara K, Maruki Y, Long X, et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 2002 ; 110 : 177-89.
- 19. Kim DH, Sarbassov DD, Ali SM, et al. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. MolCell 2003 ; 11 : 895-904.
- 20. Sarbassov D, Ali SM, Kim DH, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton.Curr Biol 2004 ; 14 : 1296-302.
- 21. Jacinto E, Loewith R, Schmidt A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol 2004 ; 6 : 1122-8.
- 22. Yonezawa K, Tokunaga C, Oshiro N, Yoshino K. Raptor, a binding partner of target of rapamycin. BiochemBiophys Res Commun 2004 ; 313 : 437-41.
- 23. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005 ; 307 : 1098-101.
- 24. Rosen ED, Walkey CJ, Puigserver P, Spiegelman BM. Transcriptional regulation of adipogenesis. Genes Dev 2000 ; 14 : 1293-307.
- 25. Bell A, Grunder L, Sorisky A. Rapamycin inhibits human adipocyte differentiation in primary culture. Obes Res 2000 ; 8 : 249-54.
- 26. Kim JE, Chen J. Regulation of peroxisome proliferator-activated receptor-gamma activity by mammalian target of rapamycin and amino acids in adipogenesis. Diabetes 2004 ; 53 : 2748-56.
- 27. El-Chaar D, Gagnon A, Sorisky A. Inhibition of insulin signaling and adipogenesis by rapamycin : effect on phosphorylation of p70 S6 kinase versus eIF4E-BP1.Int J Obes Relat Metab Disord 2004 ; 28 : 191-8.
- 28. Tsukiyama-Kohara K, Poulin F, Kohara M, et al. Adipose tissue reduction in mice lacking the translational inhibitor 4E-BP1. Nat Med 2001 ; 7 : 1128-32.
- 29. Harrington LS, Findlay GM, Lamb RF. Restraining PI3K: mTOR signalling goes back to the membrane. Trends Biochem Sci 2005 ; 30 : 35-42.
- 30. Haruta T, Uno T, Kawahara J, et al. A rapamycin-sensitive pathway down-regulates insulin signaling via phosphorylation and proteasomal degradation of insulin receptor substrate-1. Mol Endocrinol 2000 ; 14 : 783-94.
- 31. Zhang H, Cicchetti G, Onda H, et al. Loss of Tsc1/Tsc2 activates mTOR and disrupts PI3K-Akt signaling through downregulation of PDGFR. J Clin Invest 2003 ; 112 : 1223-33.
- 32. Khamzina L, Veilleux A, Bergeron S, Marette A. Increased activation of the mammalian target of rapamycin pathway in liver and skeletal muscle of obese rats: possible involvement in obesity-linked insulin resistance. Endocrinology 2005 ; 146 : 1473-81.
- 33. Um SH, Frigerio F, Watanabe M, et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 2004 ; 431 : 200-5.
- 34. Bouzakri K, Roques M, Gual P, et al. Reduced activation of phosphatidylinositol-3 kinase and increased serine 636 phosphorylation of insulin receptor substrate-1 in primary culture of skeletal muscle cells from patients with type 2 diabetes. Diabetes 2003 ; 52 : 1319-25.
List of figures
Figure 1
Contrôle de l’initiation de la traduction par 4E-BP.
La fixation du complexe eIF4F (eIF4E, eIF4G et eIF4A) à la coiffe de l’ARNm à traduire et à la sous- unité ribosomique 40S engendre un balayage du messager qui permet la localisation du codon d’initiation AUG. L’interaction de 4E-BP avec eIF4E empêche l’interaction entre eIF4E et eIF4G et, par conséquent, la formation du complexe eIF4F. Pour leur part, eIF4A et eIF4B agissent, en tant que facteurs « accessoires », en dénaturant les structures secondaires de l’ARNm. Sous sa forme liée au GTP, eIF2 interagit avec les nucléotides guanines et l’ARN de transfert méthionine (met-tARNi). Le complexe ternaire ainsi formé (eIF2·GTP·met-tARNi) se lie à la sous unité ribosomique 40S. C’est alors l’anticodon de ce met-tARNi qui reconnaît le codon d’initiation présent dans l’ARNm. ORF : origin of replication.
Figure 2
Intégration et régulation des signaux métaboliques par la voie PI3K/Akt/mTOR.
La régulation de l’activité mTOR par les facteurs de croissance fait intervenir la voie PI3K/Akt. La PI3K phosphoryle le PtdIns(4,5)P2 et produit le PtdIns(3,4,5)P3. La production de ce PtdIns(3,4,5)P3 entraîne l’activation de PDK1 qui, à son tour, active Akt. Cette activation est dépendante de PTEN. La worthmannine et le LY294002 inhibent l’activité de la PI3K. Une fois activée, cette voie de signalisation provoque la phosphorylation et l’inhibition du complexe TSC1/TSC2 par Akt, ce qui entraîne l’activation de Rheb (lié à GTP) et de mTOR. Le complexe TSC1/TSC2 joue un rôle central dans la régulation de l’activité mTOR : la présence d’acides aminés inhibe l’activité de ce complexe ; à l’inverse, une carence énergétique provoque son activation via LKB1 et AMPK. Une fois activé, le complexe mTOR/raptor/GβL, sensible à la rapamycine, relaie ces signaux vers 4E-BP et S6K. La phosphorylation de 4E-BP permet la libération d’eIF4E et la formation du complexe eIF4F nécessaire à l’initiation de la traduction. De son côté, la phosphorylation de S6K entraîne un rétrocontrôle négatif de la voie PI3K/Akt empêchant ainsi toute suractivation de la traduction cap-dépendante. L’autre complexe mTOR/rictor, insensible à la rapamycine, joue un rôle dans le réarrangement du cytosquelette.
Figure 3
Régulation de l’adipogenèse et de la sensibilité à l’insuline par mTOR.
La régulation transcriptionnelle de l’adipogenèse implique l’activation de plusieurs familles de facteurs de transcription. Ceux-ci sont chronologiquement exprimés : C/EBPβ et δ, suivis de PPARγ, qui à son tour active C/EBPα. C/EBPα, via un rétrocontôle positif sur PPARγ, maintient la différenciation adipocytaire. L’adipogenèse fait également intervenir la voie mTOR. Une fois activée, elle contrôle l’expression et l’activité de PPARγ, régulant de fait l’expression des gènes adipogéniques. Via 4E-BP, mTOR module la différenciation fonctionnelle des tissus adipeux blanc et brun. Enfin, via la S6K, mTOR est également capable de réguler la sensibilité à l’insuline.