Article body
Lorsqu’en 1906 Alois Alzheimer décrivit la maladie qui porte son nom, il rapporta la présence de lésions particulières dans le cerveau des patients : les plaques séniles extracellulaires et les dégénérescences neurofibrillaires intracellulaires. Il fallut attendre le milieu des années 1980 et les travaux respectifs de G. Glenner et J.P. Brion pour que les constituants majeurs de ces deux lésions soient caractérisés : il s’agit, d’une part, d’un peptide de 39 à 42 acides aminés, le peptide Aβ, produit lors du clivage séquentiel d’une protéine intramembranaire nommée APP (amyloid precursor protein) et, d’autre part, d’une protéine liée aux microtubules, la protéine Tau.
Les analyses génétiques menées depuis 15 ans ont montré que le déterminisme de la maladie d’Alzheimer est complexe. Dans la majorité des cas, il est polyfactoriel. Un facteur de risque génétique impliqué dans ces formes communes, l’allèle ε4 du gène de l‘apolipoprotéine E, a été identifié. Dans une minorité de cas, le déterminisme est autosomique dominant avec pénétrance complète à l’âge de 60 ans. Des mutations de type faux sens sur deux gènes, le gène APP et le gène de la préséniline 1 (PSEN1), sont responsables de la grande majorité de ces formes mendéliennes à début précoce. Les études menées au cours des années 1990 ont montré que la conséquence de ces diverses altérations génétiques était univoque. Dans tous les cas, elles s’accompagnent d’une surproduction du peptide Aβ∈42, qui est la forme la plus agrégable de ce peptide. Les mutations identifiées sur le gène APP sont essentiellement localisées au niveau des sites de clivage du peptide Aβ sur son précurseur et interfèrent avec ce clivage. La préséniline 1 est, pour sa part, un membre essentiel du complexe γ-sécrétase, responsable du clivage intramembranaire libérant le peptide Aβ à partir de son précurseur. Puisque ces mutations sont nécessaires et suffisantes pour produire à un âge très précoce (moins de 30 ans pour certaines mutations PSEN1) une maladie d’Alzheimer, il s’ensuit que le primum movens de la maladie est le dépôt de peptide Aβ et que les autres stigmates anatomo-pathologiques de la maladie sont secondaires. Cette hypothèse, dite de la cascade amyloïdergique, a été pour la première fois formulée par J. Hardy en 1995 et fait toujours l’objet d’intenses discussions [1].
Depuis 15 ans, en collaboration avec de nombreux services hospitaliers, nous analysons ces gènes dans les familles françaises concernées. Ainsi, dans 75 familles présentant des maladies d’Alzheimer à transmission autosomique dominante et à début précoce, nous avons mis en évidence 10 mutations sur l’APP et 49 sur PSEN1. Dans 12 familles où ce criblage mutationnel était resté négatif, nous avons récemment testé l’hypothèse d’une altération du dosage génique de l’APP. En effet, ce gène est situé sur le chromosome 21 et les patients trisomiques présentent à partir de 40 ans les lésions cérébrales de la maladie d’Alzheimer. Cette analyse a été réalisée au moyen d’une technique de PCR multiplex quantitative de fragments fluorescents (QMPSF) développée par notre laboratoire (Figure 1). Elle a montré que, dans cinq de ces familles, il existait une duplication du matériel génétique au niveau du locus APP [2]. Selon les familles, la taille de la duplication varie de 0,6 à 6,4 Mb et inclut de 5 à 12 gènes. Ces résultats ont été confirmés par des techniques de FISH et de PCR fluorescente quantitative au moyen de marqueurs microsatellitaires.
Figure 1
Analyse par QMPSF du dosage génique chez un sujet trisomique et un patient de la famille 028.
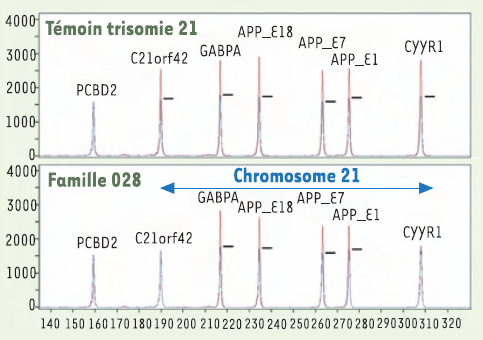
Les pics de fluorescence provenant du sujet analysé (en rouge) et d’un sujet de référence (en bleu) sont alignés à partir de l’amplicon PCBD2 situé sur le chromosome 5. Les autres amplicons sont situés sur le chromosome 21. Dans la famille 028, la duplication ne couvre ni C21orf42 ni CYYR1.
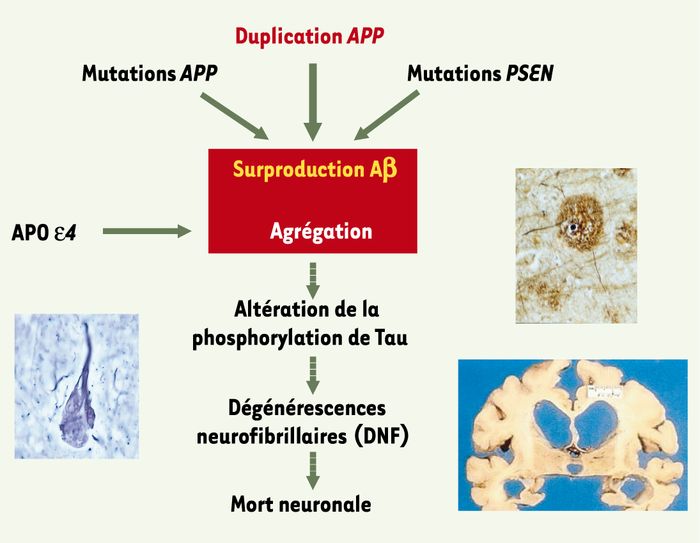
Sur le plan phénotypique, ces malades, outre une démence à début précoce, ont présenté pour certains d’entre eux des hémorragies cérébrales. L’examen neuropathologique de 5 cerveaux a montré une angiopathie amyloïde anormalement sévère. Aucun de ces patients ne présentait de manifestation clinique de trisomie 21. Au total, donc, ces résultats montrent que la duplication d’une petite région du chromosome 21 centrée sur le gène APP est suffisante pour provoquer une démence associée à une angiopathie amyloïde. Après les résultats récents concernant des duplications ou triplications de l’α-synucléine dans la maladie de Parkinson [3, 4], ils confirment que des altérations du dosage génique peuvent être à l’origine de maladies neurodégénératives provoquées par des accumulations de protéines. Enfin, ils constituent un puissant argument en faveur de l’hypothèse amyloïdergique (Figure 2) et suggèrent que d’autres mécanismes aboutissant à une expression accrue du gène APP pourraient constituer des facteurs de risque dans les formes communes de la maladie.
Figure 2
L’hypothèse de la cascade amyloïdergique.
Appendices
Références
- 1. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease : progress and problems on the road to therapeutics. Science 2002 ; 393 : 353-6.
- 2. Rovelet-Lecrux A, Hannequin D, Raux G, et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet 2006 ; 38 : 24-6.
- 3. Chartier-Harlin MC, Kachergus J, Roumier C, et al. Alpha synuclein locus duplication as a cause of familial Parkinson disease. Lancet2004 ; 364 : 1167-9.
- 4. Ibanez P, Bonnet AM, Débarges B, et al. Causal relation between alpha synuclein gene duplication and familial Parkinson disease. Lancet2004 ; 364 : 1169-71.
List of figures
Figure 1
Analyse par QMPSF du dosage génique chez un sujet trisomique et un patient de la famille 028.
Les pics de fluorescence provenant du sujet analysé (en rouge) et d’un sujet de référence (en bleu) sont alignés à partir de l’amplicon PCBD2 situé sur le chromosome 5. Les autres amplicons sont situés sur le chromosome 21. Dans la famille 028, la duplication ne couvre ni C21orf42 ni CYYR1.
Figure 2
L’hypothèse de la cascade amyloïdergique.