Abstracts
Résumé
Les canaux calciques dépendants du voltage représentent une des voies principales d’entrée du calcium dans la cellule nerveuse où ils participent activement à l’excitabilité cellulaire et aux processus moléculaires de la transmission synaptique. Ils ont, de ce fait, été depuis longtemps la cible pharmacologique d’analgésiques et ce, avant même que leur implication dans la physiologie de la nociception ait réellement été démontrée. Ces dernières années, la caractérisation moléculaire de plus en plus fine de ces canaux et de leurs sous-unités régulatrices, ainsi que la démonstration de leur implication dans les processus nociceptifs, a permis de définitivement considérer ces structures comme des cibles pharmacologiques de premier choix pour le traitement de la douleur. La recherche d’inhibiteurs spécifiques des canaux calciques dépendants du voltage laisse ainsi entrevoir le développement de nouvelles molécules analgésiques fortement prometteuses.
Summary
Voltage-dependent calcium channels represent a major pathway of calcium entry into neurons, where they participate actively to cell excitability and to the molecular processes of synaptic transmission. For that reason, they have been the direct or indirect pharmacological targets of analgesics and this long before their implication in the physiology of nociception had been demonstrated. These last years, the still more refined molecular characterization of these channels and their associated regulatory subunits and the demonstration of their implication in nociceptive processes indicates that these structures are prime pharmacological targets for the management of pain. Herein, we detail the recent breakthroughs on calcium channel structure, function and pharmacology, review the implication of calcium channels in the transmission of nociception, and evaluate their importance as targets for the treatment of pain perception. The search for specific inhibitors of voltage-dependent calcium channels appears as a prelude to the development of new promising analgesic molecules.
Article body
La définition actuelle de la douleur, établie par l’International Association for the Study of Pain®, se présente comme étant « l’expression d’une expérience sensorielle et émotionnelle désagréable liée à une lésion tissulaire existante, potentielle ou décrite en termes d’une telle lésion ». Cette définition sous-entend des mécanismes complexes d’ordre anatomophysio-pathologiques d’une part et psychologiques d’autre part, à l’origine de la sensation douloureuse [1]. Ces dernières années, la caractérisation moléculaire de plus en plus fine des voies de la nociception a précisé l’implication d’une classe particulière de canaux ioniques : les canaux calciques dépendants du voltage (CCDV). Ces structures s’avèrent être des cibles de premier choix dans le traitement de la douleur. Les événements et les voies conduisant à la sensation de la douleur sont brièvement présentés dans cet article. Cet article fait ensuite le point de manière détaillée sur les CCDV, leurs implications dans la nociception et leurs intérêts comme cible pharmacologique dans le traitement de la douleur.
Du stimulus à la sensation de douleur : un cheminement complexe
Classiquement, la nociception est décrite comme l’ensemble des fonctions de l’organisme permettant de détecter, de percevoir et de réagir à des stimuli potentiellement nocifs. À titre d’exemple, le contact avec un élément piquant se traduit rapidement par un réflexe de retrait. Il s’agit donc, avant tout, d’un mécanisme protecteur permettant à l’individu de maintenir son intégrité physique. En dehors de ce contexte, la douleur n’a pas lieu d’être. Les différentes thérapeutiques visant à l’atténuer prennent dès lors toute leur importance. On distingue deux formes principales de douleurs : la douleur de type neuropathique et celle de type nociceptif. La douleur neuropathique, la plus rare, résulte le plus souvent d’une lésion du système nerveux périphérique ou central. Il s’agit d’une douleur persistante (même après disparition de la lésion) aboutissant fréquemment à une chronicité. La douleur nociceptive provient de dommages tissulaires, autres que des tissus nerveux, et disparaît en général avec la guérison de la lésion.
Bien que la nociception soit un mécanisme hautement complexe tant d’un point de vue anatomique que moléculaire, il est possible d’en établir un schéma général (Figure 1). Le signal nociceptif prend naissance à la suite de l’activation de nocicepteurs constitués par les terminaisons libres de fibres nerveuses. On distingue les mécanonocicepteurs activés exclusivement par des stimulus intenses de nature mécanique et les nocicepteurs polymodaux, répondant non seulement à des stimulus d’origine mécanique, mais également thermique et chimique. L’activation des nocicepteurs reste un mécanisme relativement mal connu. Bien que le nocicepteur puisse être directement stimulé par l’agent nociceptif lui-même, l’implication de substances algogènes libérées en réponse à la stimulation semble être l’explication de premier choix. Parmi ces substances, on note, de manière non exhaustive, l’histamine, la sérotonine, la bradykinine, les tachykinines (substance P et neurokynine A), les prostanoïdes (prostaglandines et leucotriènes), les interleukines, les endothélines, l’ATP, mais également des éléments plus simples tels que les protons H+ ou encore certains radicaux libres comme le monoxyde d’azote [2]. Ces substances agissent sur des protéines membranaires présentes au niveau des nocicepteurs et dont l’activation est à la base même du déclenchement du signal nociceptif. L’influx nociceptif engendré est alors conduit par des fibres nerveuses de type Aδ et C en réponse à la stimulation des mécanonocicepteurs et des nocicepteurs polymodaux, respectivement. Ces neurones de premier ordre présentent tous la particularité de posséder leurs corps cellulaires dans les ganglions spinaux de la racine dorsale de la moelle épinière. Ils projettent ensuite au niveau de la corne dorsale de la moelle épinière où ils font synapses avec des neurones de second ordre. Le signal nociceptif emprunte alors les voies ascendantes via le tractus spinothalamique en direction du thalamus, puis du cortex sensoriel et du système limbique, où l’information nociceptive sera confrontée à un ensemble de processus cognitifs faisant appel, par exemple, au vécu de l’individu, à son environnement ou encore à son état émotionnel [3]. Au niveau moléculaire, la transmission de l’influx nociceptif par les neurones de premier ordre dépend très largement de diverses conductances ioniques [4] telles que des conductances sodiques, potassiques, mais également calciques engendrées par les CCDV. En contrôlant l’excitabilité cellulaire et la transmission synaptique [5], les CCDV se révèlent être à la base de la transmission de l’influx nerveux. Ainsi, ces structures apparaissent clairement comme des cibles intéressantes lorsque l’on cherche à stopper la transmission du signal nociceptif.
Figure 1
Représentation schématique des voies afférentes nociceptives.

Sont représentés quelques éléments participants au déclenchement du signal nociceptif (zoom en bas à gauche). Ce signal peut être déclenché soit par l’activation de protéines canal en réponse à un stimulus d’origine thermique ou mécanique, soit par un ensemble de molécules libérées par le tissu lésé ou sécrétées par les cellules participant au processus inflammatoire s’il a lieu. Le signal nociceptif est ensuite conduit jusque dans la corne dorsale de la moelle épinière par des neurofibres de premier ordre Aδ et C dont les propriétés structurales et fonctionnelles sont précisées dans l’encadré. La transmission du message nociceptif vers des neurones de deuxième ordre est également détaillée et met en jeux des canaux calciques dépendants du voltage. Les différentes étapes moléculaires conduisant à la transmission synaptique sont indiquées par des numéros de ① à ⑧ (zoom en haut à droite). L’information nociceptive est ensuite transmise vers les centres supérieurs via le tractus spinothalamique (figure adaptée de [2]). ASIC : acid sensing ionic channels ; P2X : récepteur ionotropique P2X ; IL-1R : récepteur de l’interleukine-1 ; B1/B2 : récepteurs des bradykinines de type 1 et 2 ; H1 : récepteur de l’histamine de type 1 ; 5-HT : récepteur de la sérotonine ; TRPV1 : transient receptor potential vanilloid 1 ; IL-1β : interleukine-1β.
Canaux calciques dépendants du voltage : diversité, structure et fonction
Les CCDV représentent une des voies majeures d’entrée de calcium dans la cellule nerveuse. Sur la base de propriétés électrophysiologiques, cette famille est subdivisée en deux classes (Tableau I) : (1) les canaux à « bas seuil d’activation » (BSA) activés par de faibles dépolarisations membranaires et (2) les canaux à « haut seuil d’activation » (HSA) activés pour de plus fortes dépolarisations membranaires. La classe des canaux BSA regroupe exclusivement les canaux de type T alors que la classe des canaux HSA comprend les canaux de type L, P/Q, N, et R [6]. Chacun de ces canaux présente des propriétés biophysiques et pharmacologiques propres. Au niveau structural, les CCDV se composent d’une sous-unité principale α1, centrée autour de sous-unités auxiliaires β, γ et α2δ (Figure 2A) dont les principales fonctions sont de moduler l’expression membranaire de la sous-unité α1(Figure 2B), son comportement biophysique ainsi que ses propriétés pharmacologiques.
Tableau I
Classification moléculaire, pharmacologique et localisation tissulaire des canaux calciques dépendants du voltage.

Figure 2
Organisation membranaire présumée des canaux calciques dépendants du voltage et des sous-unités régulatrices.

A. Les CCDV (HSA et BSA) se composent d’une sous-unité principale α1 (en bleu) formant le pore ionique. Elle se compose de quatre domaines (domaines I à IV) constitués chacun par six segments transmembranaires (S1 à S6). Ces quatre domaines sont reliés entre eux par des boucles cytoplasmiques reliant les domaines I à II (boucle I-II), II à III (boucle II-III) et III à IV (boucle III-IV). Les segments S4, riches en résidus basiques arginines et lysines, constituent le senseur de voltage. Les boucles extracellulaires et transmembranaires reliant les segments S5 et S6 (boucles P, en rouge) forment le pore ionique. L’insert représente l’organisation « tridimensionnelle » de cette sous-unité α1. Les sous-unités régulatrices des canaux HSA sont également représentées. Ces sous-unités auxiliaires modulent les propriétés biophysiques de la sous-unité α1. La sous-unité β∈(en rouge) est entièrement cytoplasmique et interagit avec la sous-unité α1via une séquence AID présente sur la boucle I-II. La sous-unité γ (en orange) est entièrement membranaire et glycosylée. La sous-unités α2δ (en vert) est essentiellement extracellulaire et ancrée à la membrane plamique (MP) par la partie δ de la protéine. Les parties α2 et δ sont reliées entre elles via deux ponts disulfures. B. La sous-unité β est responsable du ciblage fonctionnel de la sous-unité α1 à la membrane plasmique. a. Immédiatement après sa synthèse, et en absence de sous-unité β, la sous-unité α1 (en bleu) est retenue au niveau de la membrane du réticulum endoplasmique (RE) par une interaction entre la boucle cytoplasmique I-II et un élément de rétention encore non identifié (en gris). L’expression d’une sous-unité β∈(en rouge) lève cette rétention par interaction avec la boucle cytoplasmique I-II de la sous-unité α1, et cela quelle que soit l’isoforme β. À la suite de cette interaction, le complexe α1/β est adressé à la membrane plasmique. b. Il est possible d’interférer avec le processus de ciblage de la sous-unité α1 à la membrane plasmique en surexprimant une protéine chimère (en violet) constituée d’un domaine transmembranaire et de la boucle cytoplasmique I-II de la sous-unité α1. Après sa synthèse, la protéine chimère est retenue, au même titre que la sous-unité α1, au niveau de la membrane du réticulum endoplasmique via une interaction entre la boucle I-II et l’élément de rétention. La surexpression de la protéine chimère vis-à-vis de la sous-unité α1 entraîne la séquestration des sous-unités β au niveau de la membrane plasmique. Les sous-unités α1 ne sont plus ciblées correctement et restent dans la membrane du réticulum endoplasmique où elles ne sont pas fonctionnelles. AID : alpha interaction domain ; S-S : pont disulfure ; NH2 : extrémité aminoterminale ; COOH : extrémité carboxyterminale ; MP : membrane plasmique.
Bien que les CCDV présentent la particularité commune de permettre l’entrée de calcium dans la cellule, ces canaux, de par leur spécificité tissulaire, ne présentent cependant pas la même importance dans les mécanismes de transmission de l’information nociceptive.
Canaux calciques de type N
Les canaux de type N sont, pour l’essentiel, les canaux HSA les plus largement impliqués dans la nociception. Deux approches ont permis de mettre en évidence leur implication : (1) l’invalidation génique de la sous-unité α1B et (2) l’injection locale ou systémique d’inhibiteurs pharmacologiques spécifiques. L’invalidation génique de la sous-unité α1B chez l’animal témoigne de l’implication des canaux N aussi bien dans des douleurs nociceptives (thermique et inflammatoire) que neuropathiques [7, 8]. De manière intéressante, la nociception mécanique ne semble pas être affectée chez ces animaux, suggérant une certaine sélectivité dans le traitement de l’information nociceptive par ces canaux. Cette approche d’invalidation génique, bien que très informative chez l’animal, est cependant illusoire en termes de stratégie thérapeutique chez l’homme. Ainsi, l’utilisation de bloqueurs pharmacologiques spécifiques des canaux N représente sans conteste une stratégie plus rationnelle (Figure 3A). Les inhibiteurs les plus spécifiques sont l’ω-conotoxine GVIA (SNX-124) [9] et l’ω-conotoxine MVIIA (SNX-111) [10] (Tableau I). Isolés à partir du venin des mollusques marins Conus geographus et Conus magus, respectivement, ces inhibiteurs appartiennent à la classe des peptides neurotoxiques. L’administration de ces peptides par voie intrathécale chez l’animal témoigne de propriétés analgésiques intéressantes. Plus particulièrement, l’injection intrathécale d’ω-conotoxine MVIIA produit un effet antinociception [11] et anti-allodynie[1] tactile [12]. Au niveau clinique, l’injection intrathécale de ziconotide, un analogue synthétique de l’ω-conotoxine MVIIA, réalisée chez des patients cancéreux ou atteints du syndrome d’immunodéficience acquise et pour lesquels l’utilisation d’analgésiques classiques de types opiacés s’était révélé inefficace, a confirmé ses puissantes propriétés analgésiques [13] (Tableau II). Toutefois, des effets secondaires non négligeables nécessitent fréquemment l’arrêt du traitement [14]. Bien que difficile à gérer en termes de dosage, le ziconotide reste cependant une molécule intéressante. Actuellement, l’ω-conotoxine CVID (AM-336) [15], isolée à partir du venin de Conus catus, semble plus adaptée à une utilisation thérapeutique (Tableau II). En effet, ce peptide présente une toxicité amoindrie par rapport à l’ω-conotoxine MVIIA tout en conservant des propriétés analgésiques similaires [16]. Une étude récente réalisée chez l’animal montre que l’utilisation d’ω-conotoxine CVID, combinée avec le dexmédétomidine (un agoniste adrénergique également utilisé pour ses propriétés analgésiques), produit une analgésie améliorée [17]. L’utilisation combinée de différents analgésiques présente l’avantage de réduire les doses thérapeutiques, diminuant ainsi les effets indésirables liés à chacune des molécules, tout en conservant un effet analgésique satisfaisant. Toutefois, l’administration de ces molécules ne peut se faire que par voie intrathécale en raison d’un risque hypotensif trop important par d’autres voies d’administration. Actuellement, l’industrie pharmaceutique développe de nouvelles molécules visant à bloquer sélectivement les canaux N. Des dérivés de L-cystéine [18] ou de 4-aminopipéridine [19], administrés par voie orale chez l’animal, semblent posséder des propriétés analgésiques intéressantes tout en restant dénués de tout effet secondaire. Leur efficacité doit à présent être évaluée chez l’homme. Enfin, la mise en évidence récente d’une isoforme des canaux N (variant d’épissage), présente exclusivement au niveau des neurones sensitifs des ganglions spinaux, indique qu’elle puisse être une cible pharmacologique particulièrement intéressante [20]. En effet, la sélection d’inhibiteurs spécifiques de cette isoforme serait susceptible de produire, à terme, des molécules analgésiques tout aussi performantes et potentiellement administrables par voie orale en raison d’une plus grande spécificité d’action.
Tableau II
Caractéristiques analgésiques et situation clinique de différents bloqueurs de canaux calciques dépendants du voltage.
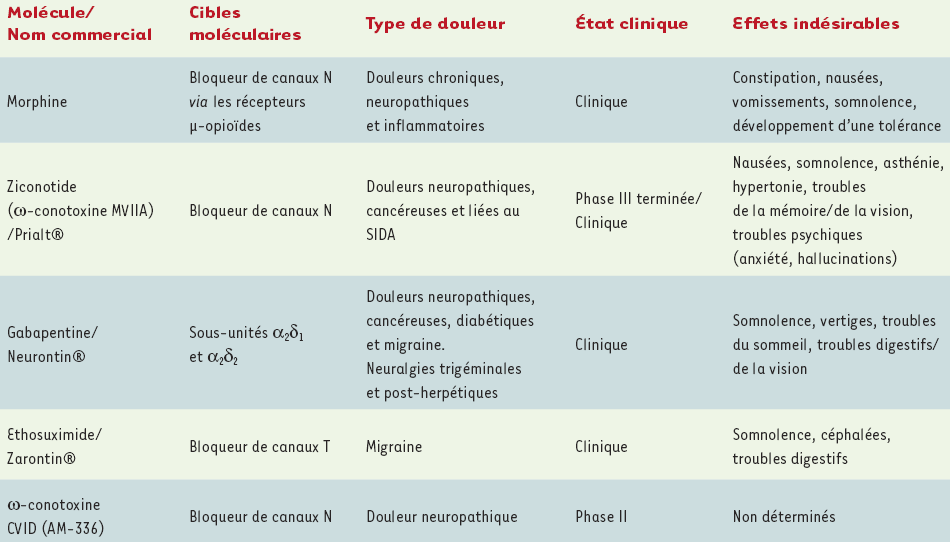
Figure 3
Représentation schématique des stratégies thérapeutiques visant à diminuer l’activité des canaux calciques dépendants du voltage.

Actuellement, deux stratégies thérapeutiques permettent de diminuer les conductances calciques soutenues par les CCDV. A. Inhibition via des bloqueurs extracellulaires. C’est le cas de l’ensemble des toxines peptidiques agissant sur les CCDV. Elles agissent en bloqueur de pore ionique en empêchant le passage des ions à travers la sous-unité α1. B. Inhibition via l’activation des récepteurs couplés aux protéines G (RCPG). Cette inhibition nécessite l’activation d’un RCPG via son ligand extracellulaire. L’activation du récepteur conduit à la dissociation du complexe Gβγ/Gα∈par échange du GDP en GTP de la sous-unité Gα. Le dimère Gβγ est alors capable de fixer directement la sous-unité α1 conduisant à une inhibition des conductances calciques. C. Représentation d’un courant calcique de type N en condition témoin (trace noire) et après activation du récepteur µ-opioïde par 10 µM de DAMGO, un analogue synthétique de la morphine. MP : membrane plasmique ; RCPG : récepteur couplé aux protéines G ; GTP : guanosine trisphosphate ; GDP : guanosine bisphosphate ; DAMGO : (D-Ala2, N-Me-Phe4, glycinol5)-Enkephalin.
Canaux N et récepteurs couplés aux protéines G
Bien que l’utilisation d’inhibiteurs agissant de manière directe sur les canaux N représente une stratégie analgésique intéressante, l’analgésique le plus ancien, encore majoritairement utilisé aujourd’hui, n’exerce pas ses effets de manière directe. Isolée en 1815, par l’Allemand Friedrich Sertüner à partir du pavot (Papaver somniferum), la morphine (Tableau II) qui doit son nom à Morphée, dieu du sommeil, a très tôt été utilisée pour ses propriétés analgésiques et calmantes. Même si les mécanismes par lesquels cet opiacé exerce ses propriétés sont restés longtemps méconnus, il est maintenant admis que la morphine agit comme un agoniste spécifique des récepteurs µ-opioïdes, lesquels appartiennent à la classe des récepteurs couplés aux protéines G (RCPG). D’un point de vue fonctionnel, l’activation des récepteurs µ-opioïdes au niveau présynaptique conduit à l’inhibition des courants calciques, notamment ceux des canaux N [21] (Figure 3B-C), et ceci grâce à la fixation du dimère Gβγ sur le canal [22, 23]. De plus, au niveau post-synaptique, l’activation des récepteurs µ-opioïdes conduit à l’activation de conductances potassiques à l’origine d’une hyperpolarisation cellulaire rendant la cellule réfractaire à toute stimulation [24]. Ainsi, en diminuant l’excitabilité cellulaire et la libération de neuromédiateurs, la morphine agit comme un puissant inhibiteur de la transmission synaptique. La caractérisation de nouveaux couples RCPG/ligands capables de moduler l’activité des canaux N se révèle être une stratégie intéressante dans la recherche de nouvelles molécules analgésiques. Le séquençage du génome humain a permis de caractériser un nombre considérable de nouveaux RCPG. À l’heure actuelle, moins de 10 % des RCPG sont utilisés comme cible thérapeutique, alors que plus de 30 % des molécules pharmaceutiques présentes sur le marché ciblent des RCPG [25]. Cela témoigne du fort potentiel thérapeutique de cette famille de molécules. Ainsi, la nociceptine (initialement nommée orphanine FQ), ligand naturel des récepteurs ORL1 (opioid receptor-like 1) présente un intérêt particulier en raison de ses effets modulateurs sur la perception de la douleur [26]. Bien que ses effets soient encore mal caractérisés, il semblerait que l’activité des canaux N soit modulée en réponse à l’activation des récepteurs ORL1 [27].
Implication des autres canaux calciques HSA
Canaux calciques de type P/Q
La caractérisation, en 1996, de différentes mutations dans le gène, présent sur le chromosome 19p13, codant pour la sous-unité α1A, a révélé l’implication des canaux P/Q dans différentes formes de migraines hémiplégiques familiales (MHF) et d’ataxie de type 2 [28]. L’étude de ces mutants a permis de mettre en évidence des modifications des propriétés biophysiques du canal conduisant à une hyperactivité neuronale des structures cérébrales impliquées dans la physiopathologie de la migraine (cortex cérébral, ganglions trijumeaux) [29]. Ainsi, les canaux P/Q représentent des cibles potentielles pour le traitement des MHF. Cependant, en raison du faible nombre de molécules disponibles pour ces canaux, ils ne sont pas les cibles de la pharmacopée actuelle. Toutefois, avec une prévalence supérieure à 20 % chez les femmes et 10 % chez les hommes, la migraine reste une affection courante, handicapante et difficilement traitable. Dans ce contexte, les canaux P/Q représentent sans aucun doute une cible pharmacologique d’intérêt à exploiter.
Canaux calciques de type R et L
Bien que les canaux N représentent les canaux HSA majoritairement impliqués dans la nociception, il n’est pas à exclure que d’autres conductances calciques aient également leur importance. L’invalidation génique, chez l’animal, des canaux R semble potentialiser les effets analgésiques de la morphine tout en diminuant l’instauration de la tolérance qui lui est associée [30]. De plus, ces canaux pourraient être impliqués dans les douleurs somatiques, même si leur implication à un niveau spinal ou supraspinal reste encore mal caractérisée [31]. Ainsi, les canaux R ne semblent présenter, pour l’instant, qu’un faible intérêt pharmacologique pour le traitement de la nociception.
Les canaux L ont également été évoqués comme pouvant participer aux mécanismes de nociception. L’injection intrapéritonéale de vérapamil, un bloqueur de canaux L, procure un effet analgésique, principalement sur des douleurs d’origine thermique et mécanique [32]. Toutefois, les évidences de leur implication dans la nociception restent encore largement fragmentaires. Néanmoins, en raison de leur sensibilité particulière aux dihydropyridines, ces canaux pourraient représenter des cibles pharmacologiques à fort intérêt thérapeutique si des preuves plus substantielles de leur implication dans des voies de signalisation nociceptive étaient faites.
Sous-unités auxiliaires : des cibles potentielles pour le traitement de la douleur
Sous-unité β
Bien que les principaux travaux se soient focalisés sur la sous-unité α?, certains résultats montrent l’implication des sous-unités régulatrices dans les mécanismes de nociception. En effet, l’invalidation génique de la sous-unité β3 chez l’animal semble conférer à ces animaux un seuil de sensibilité accru à la douleur inflammatoire [33]. Il est toutefois probable que cette sous-unité ne soit pas impliquée de manière directe dans les mécanismes de nociception, mais vraisemblablement via les canaux auxquels elle est associée. Plus particulièrement, la sous-unité β participe à la mise en place de la sous-unité α1 dans la membrane plasmique [34] (Figure 2B). Des travaux fondés sur l’utilisation d’un dominant négatif, visant à diminuer la disponibilité des sous-unités β, mettent en évidence une diminution de l’expression des canaux HSA à la membrane plasmique [35]. Cette diminution d’expression est vraisemblablement à l’origine d’une diminution de l’excitabilité cellulaire, conduisant à des effets analgésiques. En l’absence d’inhibiteurs pharmacologiques spécifiques des sous-unités β, l’utilisation d’un dominant négatif, administré localement, représente sans aucun doute une stratégie thérapeutique pertinente pour l’avenir.
Sous-unité α2δ
Il a également été relevé une augmentation de l’expression des sous-unités α2δ1 au niveau des neurones des ganglions spinaux, en réponse à une lésion du système nerveux périphérique associée au développement de douleurs allodyniques [36]. Cette surexpression des sous-unités α2δ1 n’affecte ni l’expression de la sous-unité α1B, ni celle des sous-unités β∈[37]. L’injection intrathécale d’oligonucléotides antisens, visant à diminuer le niveau d’expression des sous-unités α2δ1, confère aux animaux un effet anti-allodynie [38]. De plus, l’injection intrathécale de gabapentine (un anticonvulsif et antiépileptique) (Tableau II), capable d’interagir in vitro avec les sous-unités α2δ, diminue également les douleurs allodyniques sur ces modèles animaux. Ces données suggèrent que les sous-unités α2δ sont impliquées dans le développement des douleurs allodyniques indépendamment de leur fonction de régulation des CCDV.
Canaux calciques de type T
Ce n’est que récemment que l’injection systémique de mibéfradil, un bloqueur de canaux T (Tableau I), a permis de mettre en évidence l’implication de ces canaux dans des mécanismes de nociception [39]. Ces études réalisées chez l’animal ont mis en évidence l’implication des canaux T dans des douleurs d’origine thermique et mécanique. De même, l’injection intrapéritonéale, intrathécale ou encore locale, d’éthosuximide (un antiépileptique) (Tableau II), également bloqueur des canaux T (Tableau I), présente des propriétés analgésiques chez des modèles animaux d’hyperalgésie thermique ou mécanique [40]. Fait intéressant, l’utilisation du paclitaxel (médicament à base de taxol) classiquement utilisé dans le traitement des tumeurs pour ses propriétés antimitotiques (cette molécule bloque la réplication cellulaire en se fixant sur les microtubules qu’elle stabilise) induit chez les patients une hyperalgésie et/ou allodynie thermique et mécanique. Ces effets secondaires étant, chez le rat, complètement abolis par l’injection intrapéritonéale d’éthosuximide [41]. Ces résultats attendent d’être confirmés en clinique. Des travaux visant à développer la pharmacopée des canaux T ont permis de mettre en évidence des propriétés inhibitrices spécifiques de certains anesthésiques stéroïdiens tels que l’alphaxalone ou l’allopréggnaolone [42]. De plus, l’état redox des canaux T semble également jouer un rôle dans la modulation de la nociception. L’injection dans les aires réceptrices de L-cystéine, un agent réducteur, induit une hyperalgésie thermique qui est complètement abolie par la co-injection d’un agent oxydant comme le DTNB [43]. Ces résultats suggèrent que les sites rédox des canaux T pourraient être d’importantes cibles analgésiques. Enfin, l’implication des canaux T dans les mécanismes de nociception a été précisée très récemment par l’injection intrathécale d’oligonucléotides antisens visant à diminuer l’expression de ces canaux au niveau périphérique. Cette étude a révélé le rôle majeur de la sous-unité α1H, témoignant de l’implication sélective de ces canaux [44]. Ainsi, les canaux T représentent une classe émergente de cibles analgésiques dont la sélection d’inhibiteurs spécifiques de la sous-unité α1H est certainement une piste thérapeutique de première importance à exploiter.
Au-delà des canaux calciques dépendants du voltage…
Bien que les modulateurs des CCDV représentent des analgésiques de premier choix, ils s’intègrent toutefois dans une pharmacopée plus générale, ciblant d’autres protagonistes de la physiopathologie de la douleur. Ainsi, les cyclooxygénases (COX) impliquées dans la production des prostanoïdes sont la cible des anti-inflammatoires non stéroïdiens (AINS) tels que l’acide acétylsalicylique (aspirine) [45]. Les antidépresseurs tricycliques (ATC) sont également utilisés depuis longtemps pour leurs propriétés analgésiques. En dépit du fait que leur mécanisme d’action demeure mal connu, il est certain qu’ils exercent leurs effets analgésiques indépendamment de leurs propriétés psychogènes. En effet, en agissant comme des inhibiteurs de la recapture des monoamines (noradrénaline et sérotonine), ils participent à l’activation d’une voie nociceptive efférente inhibitrice largement dépendante de ces neuromédiateurs. Ils pourraient également moduler le système opioïde endogène ou encore agir comme des inhibiteurs des canaux ioniques. Le venlafaxine est ainsi l’ATC utilisé comme analgésique de second choix dès lors que les autres thérapeutiques se révèlent inefficaces [46].
Dans la recherche de nouvelles stratégies antidouleur, l’ensemble des canaux ioniques impliqués dans la détection, la propagation ou la transmission du signal nociceptif représente également des cibles pharmacologiques potentiellement intéressantes [47]. Dans ce contexte, l’A-317491, un inhibiteur non nucléotidique des récepteurs ionotropiques purine/pyrimidine P2X3 et P2X2/3, a démontré des propriétés analgésiques intéressantes sur des modèles animaux de neuropathies [48]. Les canaux sodiques ASIC (acidic sensing ion channels), activés par une diminution du pH extracellulaire à la suite d’un processus inflammatoire, pourraient également être des candidats intéressants si leur pharmacologie venait à se développer. En effet, seule la capsazepine, un antagoniste des canaux TRPV1, est capable de réduire l’activation des neurones sensoriels par les protons [49]. Des données plus récentes suggèrent que les AINS pourraient également moduler l’activité de ces canaux, indépendamment de leur action sur les COX [50]. Enfin, les récepteurs glutamatergiques de type NMDA (N-méthyl-D-aspartate), de par leur implication dans les phénomènes de plasticité synaptique, semblent jouer un rôle majeur dans le développement d’allodynies et d’hyperalgésies après une lésion du système nerveux périphérique [51]. Au niveau clinique, l’administration par voie intraveineuse de kétamine, un puissant antagoniste des récepteurs NMDA, a révélé des effets analgésiques très intéressants chez des patients souffrant de douleurs cancéreuses et pour lesquels la morphine s’était révélée insuffisante [52]. Toutefois, si l’implication de ces canaux ioniques dans les mécanismes moléculaires de la douleur n’est plus à démontrer, ces structures ne pourront être considérées comme de réelles cibles thérapeutiques que lorsque qu’une pharmacologie spécifique et dépourvue d’effets secondaires sera disponible.
Conclusions et perspectives
Comme nous l’avons vu en introduction, le traitement de la douleur, quel que soit son type, représente une des priorités de la thérapeutique moderne. Les canaux calciques de type N, en raison de leur implication directe dans la transmission synaptique, ont depuis longtemps été considérés comme des cibles à fort potentiel analgésique. Ils sont ainsi la cible principale de la morphine qui reste, malgré ses effets indésirables, l’analgésique le plus utilisé pour le traitement des douleurs neuropathiques. Ces dernières années, le développement de la pharmacologie des canaux N, avec la découverte de nouveaux inhibiteurs spécifiques (toxines peptidiques, petites molécules organiques), a permis d’envisager de nouvelles stratégies thérapeutiques. Un certain nombre d’essais cliniques ont ainsi pu mettre en évidence des effets analgésiques de certaines molécules dans des situations où la morphine s’était révélée inefficace. De plus, l’émergence très récente des canaux T dans les mécanismes moléculaires de la nociception ouvre de nouvelles pistes thérapeutiques. Même si la pharmacologie de ces canaux reste encore trop largement fragmentaire, nul doute que les canaux T représentent de réelles cibles pharmacologiques pour le traitement de la douleur. Ainsi, l’analgésie future repose peut-être sur l’utilisation combinée de différentes molécules agissant en synergie sur les canaux N et T. Enfin, nous avons vu que des conductances calciques sous-tendues par les autres membres de la famille des CCDV, ainsi que les sous-unités régulatrices, pourraient également avoir leur importance dans les mécanismes moléculaires de la nociception. Une caractérisation plus fine de leur implication moléculaire mérite d’être poursuivie. Les CCDV, en étant largement impliqués dans la physiologie de la nociception, représentent des cibles analgésiques à fort potentiel thérapeutiques. La meilleure compréhension de l’implication de ces canaux dans la physiologie de la nociception, ainsi que le développement d’une pharmacologie spécifique des différentes isoformes, représentent très certainement les bases actuelles qui mèneront au développement de nouvelles molécules analgésiques.
Appendices
Note
-
[1]
L’allodynie désigne une douleur déclenchée par un stimulus habituellement indolore.
Références
- 1. Craig AD. A new view of pain as a homeostatic emotion. Trends Neurosci 2003 ; 26 : 303-7.
- 2. Julius D, Basbaum AI. Molecular mechanisms of nociception. Nature 2001 ; 413 : 203-10.
- 3. Craig AD. Pain mechanisms : labeled lines versus convergence in central processing. Annu Rev Neurosci 2003 ; 26 : 1-30.
- 4. McCleskey EW, Gold MS. Ion channels of nociception. Annu Rev Physiol 1999 ; 61 : 835-56.
- 5. Meir A, Ginsburg S, Butkevich A, et al. Ion channels in presynaptic nerve terminals and control of transmitter release. Physiol Rev 1999 ; 79 : 1019-88.
- 6. Ertel EA, Campbell KP, Harpold MM, et al. Nomenclature of voltage-gated calcium channels. Neuron 2000 ; 25 : 533-5.
- 7. Hatakeyama S, Wakamori M, Ino M, et al. Differential nociceptive responses in mice lacking the alpha(1B) subunit of N-type Ca2+ channels. Neuroreport 2001 ; 12 : 2423-7.
- 8. Saegusa H, Kurihara T, Zong S, et al. Suppression of inflammatory and neuropathic pain symptoms in mice lacking the N-type Ca2+ channel. EMBO J 2001 ; 20 : 2349-56.
- 9. Kerr LM, Yoshikami D. A venom peptide with a novel presynaptic blocking action. Nature 1984 ; 308 : 282-4.
- 10. Olivera BM, Cruz LJ, de Santos V, et al. Neuronal calcium channel antagonists. Discrimination between calcium channel subtypes using omega-conotoxin from Conus magus venom. Biochemistry 1987 ; 26 : 2086-90.
- 11. Malmberg AB, Yaksh TL. Effect of continuous intrathecal infusion of omega-conopeptides, N-type calcium-channel blockers, on behavior and antinociception in the formalin and hot-plate tests in rats. Pain 1995 ; 60 : 83-90.
- 12. Chaplan SR, Pogrel JW, Yaksh TL. Role of voltage-dependent calcium channel subtypes in experimental tactile allodynia. J Pharmacol Exp Ther 1994 ; 269 : 1117-23.
- 13. Staats PS, Yearwood T, Charapata SG, et al. Intrathecal ziconotide in the treatment of refractory pain in patients with cancer or AIDS : a randomized controlled trial. JAMA 2004 ; 291 : 63-70.
- 14. Penn RD, Paice JA. Adverse effects associated with the intrathecal administration of ziconotide. Pain 2000 ; 85 : 291-6.
- 15. Adams DJ, Smith AB, Schroeder CI, et al. Omega-conotoxin CVID inhibits a pharmacologically distinct voltage-sensitive calcium channel associated with transmitter release from preganglionic nerve terminals. J Biol Chem 2003 ; 278 : 4057-62.
- 16. Smith MT, Cabot PJ, Ross FB, et al. The novel N-type calcium channel blocker, AM336, produces potent dose-dependent antinociception after intrathecal dosing in rats and inhibits substance P release in rat spinal cord slices. Pain 2002 ; 96 : 119-27.
- 17. Blake DW, Scott DA, Angus JA, Wright CE. Synergy between intrathecal omega-conotoxin CVID and dexmedetomidine to attenuate mechanical hypersensitivity in the rat. Eur J Pharmacol 2005 ; 506 : 221-7.
- 18. Seko T, Kato M, Kohno H, et al. Structure-activity study of L-cysteine-based N-type calcium channel blockers : optimization of N- and C-terminal substituents. Bioorg Med Chem Lett 2002 ; 12 : 915-8.
- 19. Teodori E, Baldi E, Dei S, et al. Design, synthesis, and preliminary pharmacological evaluation of 4-aminopiperidine derivatives as N-type calcium channel blockers active on pain and neuropathic pain. J Med Chem 2004 ; 47 : 6070-81.
- 20. Bell TJ, Thaler C, Castiglioni AJ, et al. Cell-specific alternative splicing increases calcium channel current density in the pain pathway. Neuron 2004 ; 41 : 127-38.
- 21. Seward E, Hammond C, Henderson G. Mu-opioid-receptor-mediated inhibition of the N-type calcium-channel current. Proc Biol Sci 1991 ; 244 : 129-35.
- 22. De Waard M, Liu H, Walker D, et al. Direct binding of G-protein betagamma complex to voltage-dependent calcium channels. Nature 1997 ; 385 : 446-50.
- 23. Zamponi GW, Bourinet E, Nelson D, et al. Crosstalk between G proteins and protein kinase C mediated by the calcium channel alpha1 subunit. Nature 1997 ; 385 : 442-6.
- 24. Marker CL, Lujan R, Loh HH, Wickman K. Spinal G-protein-gated potassium channels contribute in a dose-dependent manner to the analgesic effect of mu- and delta- but not kappa-opioids. J Neurosci 2005 ; 25 : 3551-9.
- 25. Wise A, Gearing K, Rees S. Target validation of G-protein coupled receptors. Drug Discov Today 2002 ; 7 : 235-46.
- 26. Meunier JC. Nociceptin/orphanin FQ and the opioid receptor-like ORL1 receptor. Eur J Pharmacol 1997 ; 340 : 1-15.
- 27. Beedle AM, McRory JE, Poirot O, et al. Agonist-independent modulation of N-type calcium channels by ORL1 receptors. Nat Neurosci 2004 ; 7 : 118-25.
- 28. Ophoff RA, Terwindt GM, Vergouwe MN, et al. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell 1996 ; 87 : 543-52.
- 29. Pietrobon D, Striessnig J. Neurobiology of migraine. Nat Rev Neurosci 2003 ; 4 : 386-98.
- 30. Yokoyama K, Kurihara T, Saegusa H, et al. Blocking the R-type (Cav2.3) Ca2+ channel enhanced morphine analgesia and reduced morphine tolerance. Eur J Neurosci 2004 ; 20 : 3516-9.
- 31. Saegusa H, Kurihara T, Zong S, et al. Altered pain responses in mice lacking alpha 1E subunit of the voltage-dependent Ca2+ channel. Proc Natl Acad Sci USA 2000 ; 97 : 6132-7.
- 32. Todorovic SM, Pathirathna S, Meyenburg A, Jevtovic-Todorovic V. Mechanical and thermal anti-nociception in rats after systemic administration of verapamil. Neurosci Lett 2004 ; 360 : 57-60.
- 33. Murakami M, Fleischmann B, De Felipe C, et al. Pain perception in mice lacking the beta3 subunit of voltage-activated calcium channels. J Biol Chem 2002 ; 277 : 40342-51.
- 34. Bichet D, Cornet V, Geib S, et al. The I-II loop of the Ca2+ channel alpha1 subunit contains an endoplasmic reticulum retention signal antagonized by the beta subunit. Neuron 2000 ; 25 : 177-90.
- 35. Cuchillo-Ibanez I, Aldea M, Brocard J, et al. Inhibition of voltage-gated calcium channels by sequestration of beta subunits. Biochem Biophys Res Commun 2003 ; 311 : 1000-7.
- 36. Newton RA, Bingham S, Case PC, et al. Dorsal root ganglion neurons show increased expression of the calcium channel alpha2delta-1 subunit following partial sciatic nerve injury. Brain Res Mol Brain Res 2001 ; 95 : 1-8.
- 37. Luo ZD, Chaplan SR, Higuera ES, et al. Upregulation of dorsal root ganglion (alpha)2(delta) calcium channel subunit and its correlation with allodynia in spinal nerve-injured rats. J Neurosci 2001 ; 21 : 1868-75.
- 38. Li CY, Song YH, Higuera ES, Luo ZD. Spinal dorsal horn calcium channel alpha2delta-1 subunit upregulation contributes to peripheral nerve injury-induced tactile allodynia. J Neurosci 2004 ; 24 : 8494-9.
- 39. Todorovic SM, Meyenburg A, Jevtovic-Todorovic V. Mechanical and thermal antinociception in rats following systemic administration of mibefradil, a T-type calcium channel blocker. Brain Res 2002 ; 951 : 336-40.
- 40. Dogrul A, Gardell LR, Ossipov MH, et al. Reversal of experimental neuropathic pain by T-type calcium channel blockers. Pain 2003 ; 105 : 159-68.
- 41. Flatters SJ, Bennett GJ. Ethosuximide reverses paclitaxel- and vincristine-induced painful peripheral neuropathy. Pain 2004 ; 109 : 150-61.
- 42. Todorovic SM, Pathirathna S, Brimelow BC, et al. 5beta-reduced neuroactive steroids are novel voltage-dependent blockers of T-type Ca2+ channels in rat sensory neurons in vitro and potent peripheral analgesics in vivo. Mol Pharmacol 2004 ; 66 : 1223-35.
- 43. Todorovic SM, Meyenburg A, Jevtovic-Todorovic V. Redox modulation of peripheral T-type Ca2+ channels in vivo : alteration of nerve injury-induced thermal hyperalgesia. Pain 2004 ; 109 : 328-39.
- 44. Bourinet E, Alloui A, Monteil A, et al. Silencing of the Ca(v)3.2 T-type calcium channel gene in sensory neurons demonstrates its major role in nociception. EMBO J 2005 ; 24 : 315-24.
- 45. Burian M, Geisslinger G. COX-dependent mechanisms involved in the antinociceptive action of NSAIDs at central and peripheral sites. Pharmacol Ther 2005 ; 107 : 139-54.
- 46. Coluzzi F, Mattia C. Mechanism-based treatment in chronic neuropathic pain : the role of antidepressants. Curr Pharm Des 2005 ; 11 : 2945-60.
- 47. Eglen RM, Hunter JC, Dray A. Ions in the fire : recent ion-channel research and approaches to pain therapy. Trends Pharmacol Sci 1999 ; 20 : 337-42.
- 48. Jarvis MF, Burgard EC, McGaraughty S, et al. A-317491, a novel potent and selective non-nucleotide antagonist of P2X3 and P2X2/3 receptors, reduces chronic inflammatory and neuropathic pain in the rat. Proc Natl Acad Sci USA 2002 ; 99 : 17179-84.
- 49. Vyklicky L, Knotkova-Urbancova H, Vitaskova Z, et al. Inflammatory mediators at acidic pH activate capsaicin receptors in cultured sensory neurons from newborn rats. J Neurophysiol 1998 ; 79 : 670-6.
- 50. Voilley N. Acid-sensing ion channels (ASICs) : new targets for the analgesic effects of non-steroid anti-inflammatory drugs (NSAIDs). Curr Drug Targets Inflamm Allergy 2004 ; 3 : 71-9.
- 51. Wilson JA, Garry EM, Anderson HA, et al. NMDA receptor antagonist treatment at the time of nerve injury prevents injury-induced changes in spinal NR1 and NR2B subunit expression and increases the sensitivity of residual pain behaviours to subsequently administered NMDA receptor antagonists. Pain 2005 ; 117 : 421-32.
- 52. Lossignol DA, Obiols-Portis M, Body JJ. Successful use of ketamine for intractable cancer pain. Support Care Cancer 2005 ; 13 : 188-93.
List of figures
Figure 1
Représentation schématique des voies afférentes nociceptives.
Sont représentés quelques éléments participants au déclenchement du signal nociceptif (zoom en bas à gauche). Ce signal peut être déclenché soit par l’activation de protéines canal en réponse à un stimulus d’origine thermique ou mécanique, soit par un ensemble de molécules libérées par le tissu lésé ou sécrétées par les cellules participant au processus inflammatoire s’il a lieu. Le signal nociceptif est ensuite conduit jusque dans la corne dorsale de la moelle épinière par des neurofibres de premier ordre Aδ et C dont les propriétés structurales et fonctionnelles sont précisées dans l’encadré. La transmission du message nociceptif vers des neurones de deuxième ordre est également détaillée et met en jeux des canaux calciques dépendants du voltage. Les différentes étapes moléculaires conduisant à la transmission synaptique sont indiquées par des numéros de ① à ⑧ (zoom en haut à droite). L’information nociceptive est ensuite transmise vers les centres supérieurs via le tractus spinothalamique (figure adaptée de [2]). ASIC : acid sensing ionic channels ; P2X : récepteur ionotropique P2X ; IL-1R : récepteur de l’interleukine-1 ; B1/B2 : récepteurs des bradykinines de type 1 et 2 ; H1 : récepteur de l’histamine de type 1 ; 5-HT : récepteur de la sérotonine ; TRPV1 : transient receptor potential vanilloid 1 ; IL-1β : interleukine-1β.
Figure 2
Organisation membranaire présumée des canaux calciques dépendants du voltage et des sous-unités régulatrices.
A. Les CCDV (HSA et BSA) se composent d’une sous-unité principale α1 (en bleu) formant le pore ionique. Elle se compose de quatre domaines (domaines I à IV) constitués chacun par six segments transmembranaires (S1 à S6). Ces quatre domaines sont reliés entre eux par des boucles cytoplasmiques reliant les domaines I à II (boucle I-II), II à III (boucle II-III) et III à IV (boucle III-IV). Les segments S4, riches en résidus basiques arginines et lysines, constituent le senseur de voltage. Les boucles extracellulaires et transmembranaires reliant les segments S5 et S6 (boucles P, en rouge) forment le pore ionique. L’insert représente l’organisation « tridimensionnelle » de cette sous-unité α1. Les sous-unités régulatrices des canaux HSA sont également représentées. Ces sous-unités auxiliaires modulent les propriétés biophysiques de la sous-unité α1. La sous-unité β∈(en rouge) est entièrement cytoplasmique et interagit avec la sous-unité α1via une séquence AID présente sur la boucle I-II. La sous-unité γ (en orange) est entièrement membranaire et glycosylée. La sous-unités α2δ (en vert) est essentiellement extracellulaire et ancrée à la membrane plamique (MP) par la partie δ de la protéine. Les parties α2 et δ sont reliées entre elles via deux ponts disulfures. B. La sous-unité β est responsable du ciblage fonctionnel de la sous-unité α1 à la membrane plasmique. a. Immédiatement après sa synthèse, et en absence de sous-unité β, la sous-unité α1 (en bleu) est retenue au niveau de la membrane du réticulum endoplasmique (RE) par une interaction entre la boucle cytoplasmique I-II et un élément de rétention encore non identifié (en gris). L’expression d’une sous-unité β∈(en rouge) lève cette rétention par interaction avec la boucle cytoplasmique I-II de la sous-unité α1, et cela quelle que soit l’isoforme β. À la suite de cette interaction, le complexe α1/β est adressé à la membrane plasmique. b. Il est possible d’interférer avec le processus de ciblage de la sous-unité α1 à la membrane plasmique en surexprimant une protéine chimère (en violet) constituée d’un domaine transmembranaire et de la boucle cytoplasmique I-II de la sous-unité α1. Après sa synthèse, la protéine chimère est retenue, au même titre que la sous-unité α1, au niveau de la membrane du réticulum endoplasmique via une interaction entre la boucle I-II et l’élément de rétention. La surexpression de la protéine chimère vis-à-vis de la sous-unité α1 entraîne la séquestration des sous-unités β au niveau de la membrane plasmique. Les sous-unités α1 ne sont plus ciblées correctement et restent dans la membrane du réticulum endoplasmique où elles ne sont pas fonctionnelles. AID : alpha interaction domain ; S-S : pont disulfure ; NH2 : extrémité aminoterminale ; COOH : extrémité carboxyterminale ; MP : membrane plasmique.
Figure 3
Représentation schématique des stratégies thérapeutiques visant à diminuer l’activité des canaux calciques dépendants du voltage.
Actuellement, deux stratégies thérapeutiques permettent de diminuer les conductances calciques soutenues par les CCDV. A. Inhibition via des bloqueurs extracellulaires. C’est le cas de l’ensemble des toxines peptidiques agissant sur les CCDV. Elles agissent en bloqueur de pore ionique en empêchant le passage des ions à travers la sous-unité α1. B. Inhibition via l’activation des récepteurs couplés aux protéines G (RCPG). Cette inhibition nécessite l’activation d’un RCPG via son ligand extracellulaire. L’activation du récepteur conduit à la dissociation du complexe Gβγ/Gα∈par échange du GDP en GTP de la sous-unité Gα. Le dimère Gβγ est alors capable de fixer directement la sous-unité α1 conduisant à une inhibition des conductances calciques. C. Représentation d’un courant calcique de type N en condition témoin (trace noire) et après activation du récepteur µ-opioïde par 10 µM de DAMGO, un analogue synthétique de la morphine. MP : membrane plasmique ; RCPG : récepteur couplé aux protéines G ; GTP : guanosine trisphosphate ; GDP : guanosine bisphosphate ; DAMGO : (D-Ala2, N-Me-Phe4, glycinol5)-Enkephalin.
List of tables
Tableau I
Classification moléculaire, pharmacologique et localisation tissulaire des canaux calciques dépendants du voltage.
Tableau II
Caractéristiques analgésiques et situation clinique de différents bloqueurs de canaux calciques dépendants du voltage.