Article body
C’est grâce aux conclusions du rapport « les orphelins de la santé » écrit par Annie Wolf en 1994 à la demande de Philippe Lazar que le Ministère des Affaires Sociales créa la Mission des Médicaments orphelins. Celle-ci fut dotée de deux objectifs, promouvoir une politique européenne de développement de médicaments orphelins et accompagner ce projet européen par des initiatives nationales. Plusieurs années d’efforts aboutirent à l’adoption, le 16 décembre 1999, par le parlement européen et le conseil, du règlement européen sur les médicaments orphelins [1]. Soulignons que l’Europe n’est pas pionnière en la matière : ce sont en effet les États-Unis qui légiférèrent les premiers en adoptant l’Orphan Drug Act [2, 3] dès 1983, suivis par le Japon en 1993, Taiwan en 1997 et l’Australie en 1998. S’il existe quelques différences selon les législations, l’objectif et les moyens utilisés sont bien les mêmes : inciter les industries pharmaceutiques, en leur accordant un certain nombre d’avantages, à développer des médicaments pour le traitement des maladies rares, dits médicaments orphelins.
Précisons tout d’abord ce que l’on entend par médicament « orphelin ». Dans le cadre de cette loi, sont désignés « orphelins » les médicaments destinés à la prévention, au diagnostic ou au traitement d’une affection entraînant une menace pour la vie ou une invalidité chronique ne touchant pas plus de 5 personnes pour 10 000 dans l’Union Européenne. Il s’agit donc ici d’une définition épidémiologique fondée sur la notion de prévalence de la maladie. Ainsi, aux États-Unis, une maladie est dite rare lorsqu’elle touche moins de 200 000 personnes (soit 7,5 personnes pour 10 000), au Japon le seuil de prévalence est de 4 pour 10 000 tandis qu’il n’est que de 1 pour 10 000 en Australie. Notons qu’à ces critères épidémiologiques s’ajoute une définition économique, un médicament peut en effet être désigné « orphelin » s’il s’applique à une maladie très grave et/ou invalidante, et qu’il est peu probable qu’en l’absence de mesures d’incitation, la commercialisation de ce médicament soit suffisamment rentable. Une telle définition s’applique en théorie aux médicaments pour les maladies dites « négligées » (neglected diseases) correspondant aux maladies, le plus souvent infectieuses, des pays pauvres. Nous verrons qu’en fait bien peu de médicaments ont été désignés « orphelins » selon cette définition.
En Europe, la désignation de « médicaments orphelins » est accordée par l’agence Européenne des Médicaments (EMEA) après avoir obtenu l’avis d’une commission ad hoc créée en son sein. Il s’agit du Comité des Médicaments Orphelins (COMP) dont une des grandes caractéristiques est d’associer spécialistes du médicament et représentants des associations de malades. La désignation peut se faire à tous les stades de développement d’un médicament, elle n’équivaut en aucun cas à une autorisation d’utilisation, ni bien sûr à une recommandation d’utilisation, mais permet de bénéficier d’un certain nombre de mesures incitatives. Les médicaments désignés orphelins ont en effet un accès direct à une procédure centralisée d’autorisation de mise sur le marché passant par l’EMEA et bénéficient d’une exclusivité commerciale dans la Communauté Européenne de dix ans après l’octroi de l’AMM. Par ailleurs, les promoteurs peuvent bénéficier de réductions ou d’exonérations des droits d’enregistrement et d’une assistance à l’élaboration de protocoles et au développement, augmentant ainsi leurs chances d’obtenir l’AMM et d’en hâter son obtention.
Après cinq ans de fonctionnement, un bilan globalement positif
Plus de 500 dossiers ont été déposés à l’EMEA ; 315 médicaments ont été désignés orphelins, parmi lesquels 44 ont fait l’objet d’une demande d’AMM dont 22 ont abouti à une AMM par procédure centralisée (Tableaux I et II).
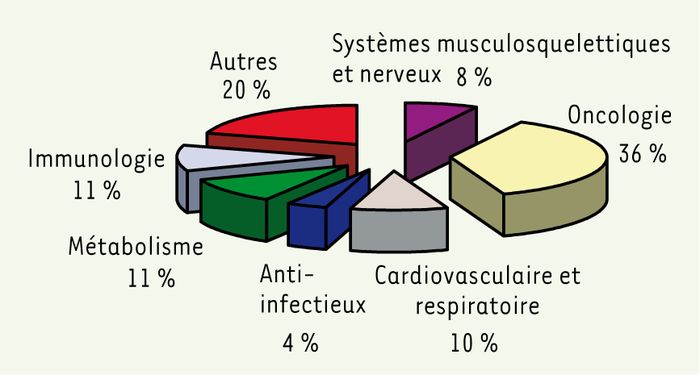
Le bilan effectué jusqu’en avril 2005 (portant sur 458 dossiers) [4] montre que 90 % des produits désignés concernent des maladies dont la prévalence est inférieure à 3 pour 10 000, parmi lesquels 43 % de prévalence inférieure à 1 pour 10 000. Parmi les opinions positives, le domaine de l’oncologie est de loin le plus représenté (38 %), en particulier les lymphomes, les leucémies et les gliomes (6 %) suivi de l’immunologie (11 %) et des maladies métaboliques (11 %) (Figure 1). Viennent ensuite les pathologies pulmonaires et respiratoires (10 %), représentées en grande partie par la mucoviscidose, et les maladies neurologiques et neuromusculaires (8 %). Les pathologies infectieuses ne représentent que 4 % des désignations orphelines, et moins de dix produits concernent les maladies dites « négligées » (lèpre, leishmaniose viscérale, tuberculose…).
Figure 1
Distribution des désignations orphelines selon les spécialités médicales.
Plus de la moitié des produits désignés sont des produits innovants et 20 % sont issus des biotechnologies, thérapie génique, thérapie antisens, anticorps monoclonaux et produits recombinants. Notons également que 46 % des produits sont destinés aux adultes, 43 % à un usage pédiatrique et 11 % à un usage mixte. Enfin, la contribution financière de l’Union Européenne a été d’environ 12 millions Ð depuis le début de la législation, la moitié pour l’assistance aux protocoles (80 procédures on été engagées) (Figure 2).
Figure 2
Contribution financière de l’Union Européenne.
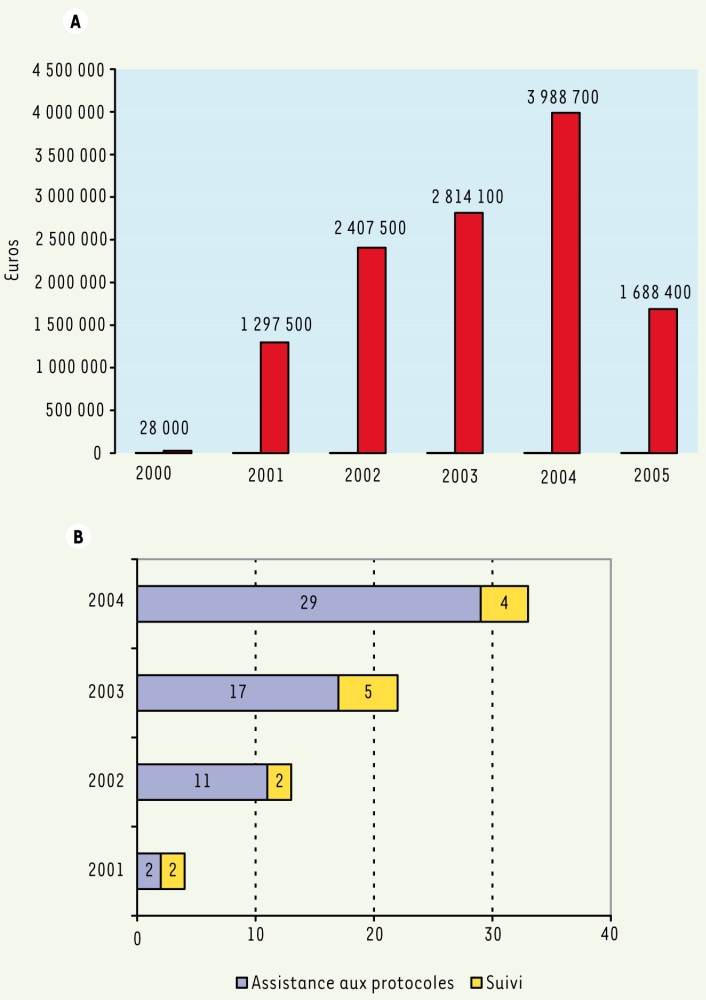
A. Distribution par année des réductions de charges. B. Nombre de procédures d’assistance aux protocoles engagées par année.
Tableau I
Produits orphelins ayant bénéficié d’une AMM européenne par procédure centralisée.
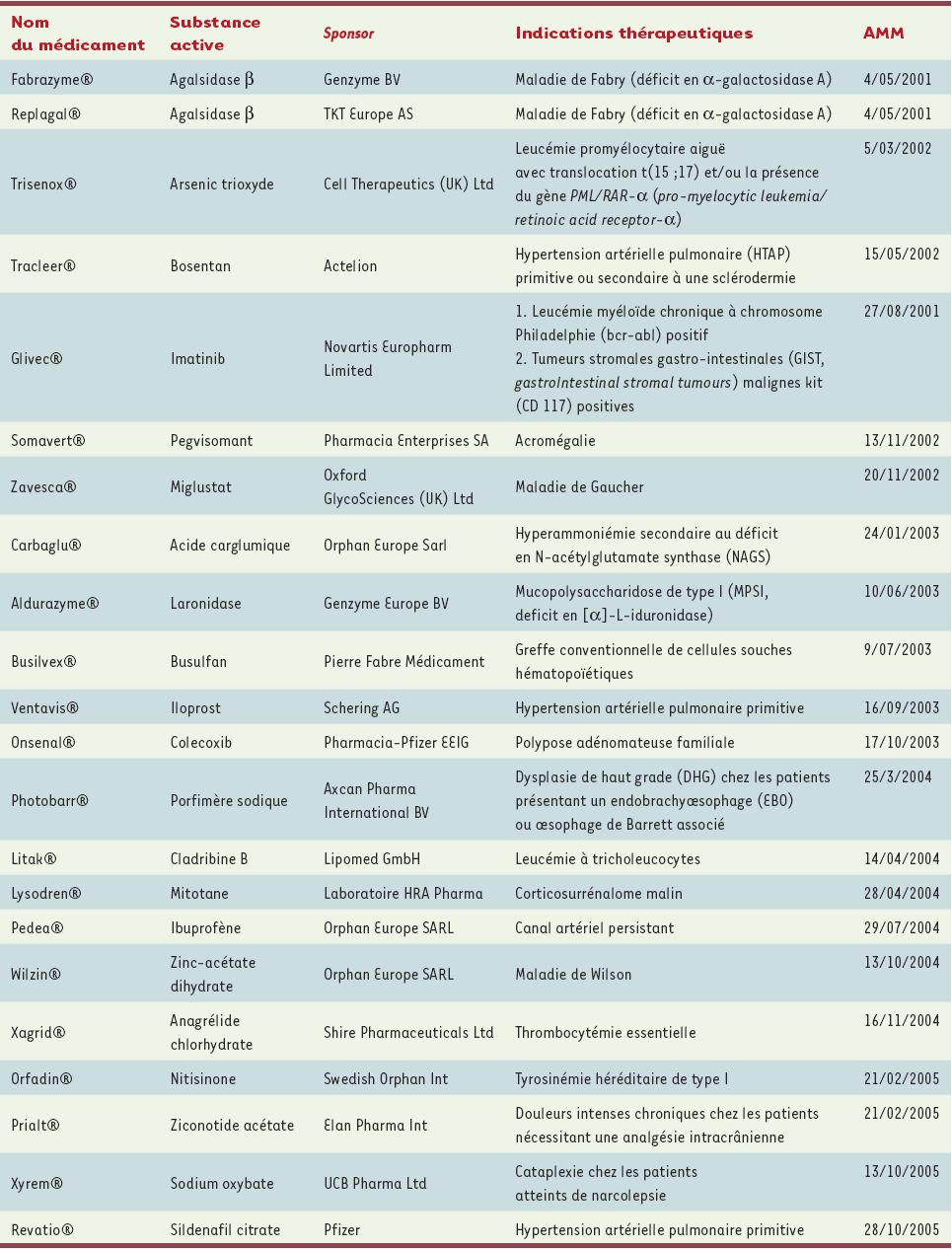
AMM : autorisation de mise sur le marché.
Tableau II
Désignation des médicaments orphelins depuis la mise en place de la réglementation européenne.
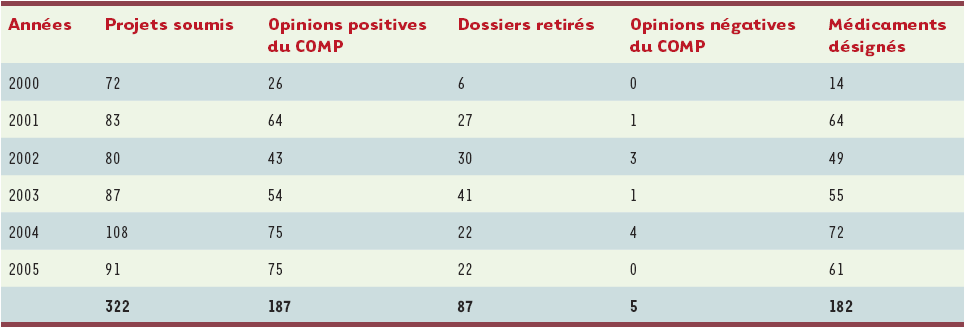
COMP : comité des médicaments orphelins (mise à jour d’octobre 2005).
Ainsi, la législation mise en place par l’Europe a été, à l’évidence, bénéfique pour le développement et la commercialisation des médicaments orphelins. Les résultats sont d’ailleurs assez proches de ceux des États-Unis où environ 1 500 produits orphelins ont été désignés et 270 commercialisés, 22 ans après l’adoption de l’Orphan Drug Act (d’après [5], données d’avril 2005).
La persistance de difficultés
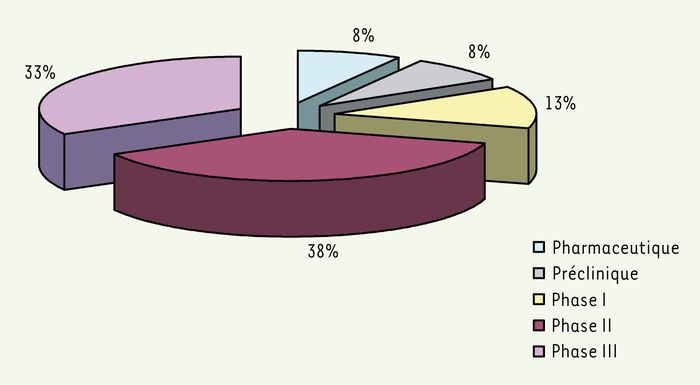
De nombreuses difficultés persistent néanmoins. Ainsi, contrairement à ce qui a été mis en place aux États-Unis, il n’existe actuellement en Europe aucun programme spécifique destiné à soutenir les essais cliniques pour les produits orphelins. La désignation pouvant se faire à tous les stades de recherche et développement (Figure 3), y compris pré-clinique, cette absence de soutien aux essais cliniques qui sont le plus souvent très onéreux, risque à terme de ralentir voire limiter le passage des produits « désignés orphelins » à leur autorisation. Un autre point important est le prix élevé de certains de ces médicaments avec un coût annuel de traitement pouvant atteindre plusieurs centaines de milliers d’€ notamment pour les thérapies de remplacement enzymatiques. Pour certains, ces prix élevés seraient une conséquence directe du monopole dont bénéficient les compagnies pharmaceutiques. Pour ces dernières, il s’agit en revanche d’une conséquence du faible nombre de malades concernés et de la nécessité d’avoir un retour sur investissement suffisant alors que le coût du développement de ces médicaments est très élevé. Notons également que le chapitre 8.2 de la réglementation, qui introduit une réserve pour limiter la période d’exclusivité à 6 ans, si le produit est suffisamment « profitable », maintient un certain degré d’ambiguïté qu’il est d’autant plus important de lever que les premiers médicaments orphelins arrivent bientôt au terme de leur 5e année de mise sur le marché. Enfin, le délai d’accès aux médicaments orphelins autorisés peut également poser problème car l’obtention d’une AMM européenne ne signifie en effet pas que ce médicament est réellement disponible dans les 25 pays de l’Union européenne. Une enquête menée en décembre 2004 par EURORDIS (European organisation for rare disorders) sur la disponibilité de 12 médicaments après au minimum un an d’AMM montrait en effet une très grande disparité selon les pays, les 12 médicaments n’étant tous disponibles que dans un seul pays, le Danemark [6].
Figure 3
État de développement des produits désignés.
La législation sur les médicaments orphelins fait des petits…
S'inspirant en grande partie de la réglementation Européenne sur les médicaments orphelins et de la législation américaine, une nouvelle réglementation, sur les médicaments pédiatriques, devrait être adoptée très prochainement par le Conseil Européen. Elle vise à favoriser la recherche, le développement et l'autorisation de médicaments à usage pédiatrique. Un comité des médicaments pédiatriques sera ainsi mis en place au sein de l'EMEA. Une des mesures incitatives phare sera l'obtention d'une prolongation de 6 mois du monopole de commercialisation pour les médicaments qui respecteront cette nouvelle réglementation. Quant aux maladies rares, elles sont bien évidemment directement concernées par ce texte, et il est d'ailleurs proposé de porter de dix à douze ans la période d'exclusivité commerciale pour les médicaments orphelins à usage pédiatrique.
Appendices
Références
- 1. Regulation (EC) n° 141/2000 of the European Parliament and of the Council of 16 December 1999 on orphan medicinal products. Official Journal of the European Communities 22 janvier 2000.
- 2. The orphan drug act. US Food and Drug Administration. www.fda.gov/orphan/oda.htm
- 3. Haffner ME, Whitley J, Moses M. Two decades of orphan product development. Nat Rev Drug Discov 2002 ; 1 : 821-5.
- 4. COMP report to the commission in relation to article 10 of regulation 141/2000 on orphan medicinal products. http://www.emeaeu.int/index/indexh1.htm
- 5. Rinaldi A. Adopting an Orphan. EMBO Rep 2005 ; 6 : 507-10.
- 6. Héron E. Les médicaments orphelins en Europe. Med Sci(Paris) 2005 ; 21 (n° spécial) : 66-8.
List of figures
Figure 1
Distribution des désignations orphelines selon les spécialités médicales.
Figure 2
Contribution financière de l’Union Européenne.
A. Distribution par année des réductions de charges. B. Nombre de procédures d’assistance aux protocoles engagées par année.
Figure 3
État de développement des produits désignés.
List of tables
Tableau I
Produits orphelins ayant bénéficié d’une AMM européenne par procédure centralisée.
AMM : autorisation de mise sur le marché.
Tableau II
Désignation des médicaments orphelins depuis la mise en place de la réglementation européenne.
COMP : comité des médicaments orphelins (mise à jour d’octobre 2005).